生物质转化及生物质油精制的研究进展
2010-06-05王勇,邹献武,秦特夫
生物质能是太阳能以化学能形式储存在生物质中的能量形式,是以生物质为载体的可再生能源。我国生物质资源十分丰富,资源总量超过30亿t·a-1,相当于10亿t·a-1油当量,约为我国目前石油消耗量的3倍。其中,每年仅农作物秸秆和农副产品谷壳等就有7亿多吨,除30%用作饲料、肥料和工业原料外,约60%可以作为能源使用[1]。由于化石能源的逐渐枯竭及其不可再生的严峻形势,使得开发可再生能源、更新能源结构、维持人类可持续发展的研究格外引人注目,生物质转化的研究也因此受到重视[2,3]。生物质油是生物质在非热力学平衡条件下分解反应的产物,是由木质素、纤维素和半纤维素通过解聚过程得到的,成分复杂且对热不稳定,是初级的液体燃料[4,5]。因此,需检测了解生物质油性质,对生物质油进行精制以改善其品质,从而满足生物质油作为燃料油的要求。
作者在此详细介绍了两种生物质转化技术——生物质热解和生物质液化,比较了这两种转化技术所得生物质油的化学组成,评述了生物质油精制技术。
1 生物质转化技术
生物质的转化技术很多,主要有直接燃烧、生物发酵和热化学转化等[6~9]。目前,生物质制备液体燃料的方法见图1。
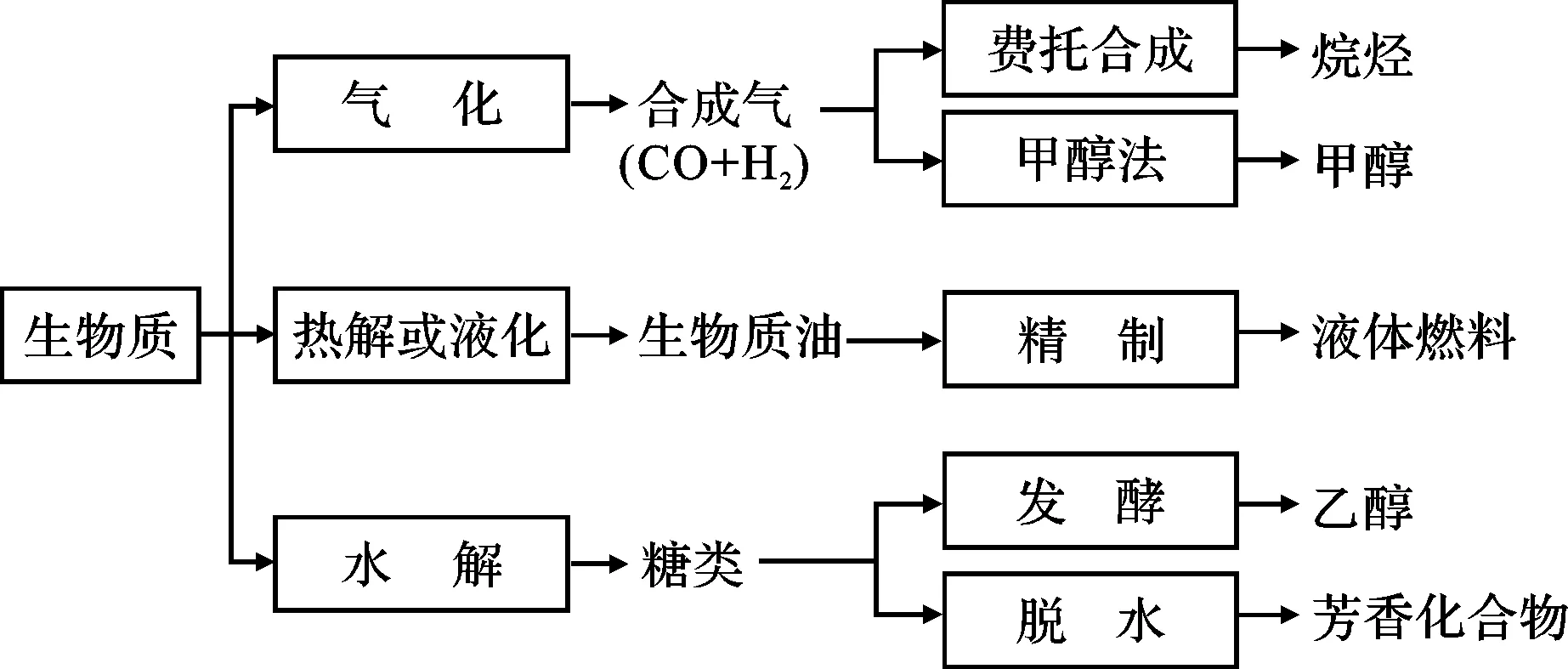
图1 生物质制备液体燃料的方法
生物质热解和生物质液化是制备生物质油最常用的两种方法。生物质热解是指在隔绝空气或通入少量空气的条件下,利用热能切断生物质大分子的化学键,使之转变为小分子物质的过程。从对生物质的加热速率和完成反应时间来看,生物质热解工艺基本上可以分为两种类型:一是慢速热解;二是快速热解。在快速热解中,当完成反应时间极短(<0.5 s)时,又被称为闪速热解。具体工艺参数比较见表1。

表1 不同热解类型主要运行参数[10]
快速热解主要产物是生物质油。慢速热解的产物以木炭为主,产炭率最高可达35%。而闪速热解的产物以可燃性气体为主。
生物质液化的研究始于20世纪70年代,近40年来,生物质液化的研究主要集中在液化工艺的探索、液化机理的研究和液化产物的综合利用这三个方面。目前采用的主要液化工艺还是用苯酚或多元醇作溶剂,在酸性、碱性或金属盐存在的条件下,将未经任何化学处理的生物质进行液化。有学者用一元醇作溶剂、浓硫酸作催化剂,在密闭的条件下对木粉进行液化[11],液化率最高能达到80%,但该液化法对木质素的液化效果不理想,产物中以木质素降解产物为主的重质组分含量较高,还需要进一步处理。还有学者将超临界流体技术应用到生物质液化中,Chumpoo等[12]研究了在超临界乙醇条件下对甘蔗渣的液化,甘蔗渣转化率可达99.9%,液化油得率最高可达73.8%,所得液化油的热值为26.8 MJ·kg-1,虽然比甘蔗渣的热值(14.8 MJ·kg-1)有很大提高,但相比于汽油的热值(40 MJ·kg-1),超临界流体液化油还需要进一步精制,以提高热值。与生物质热解技术相比,生物质液化技术可以生产出物理稳定性和化学稳定性都更好的液体产品。
2 生物质油性质及化学组成
生物质油主要由一些相对分子量较大的有机化合物组成,是非常复杂的混合物,其物理化学性质取决于生物质原料的种类、裂解方法和产物分离效率等因素。不同裂解方法制备的生物质油与普通石油在性质上有明显区别,生物质快速热解油、生物质液化油和石化重油的性质见表2[13]。
由表2可知,生物质快速热解油相比于传统的石化重油有更高的含氧量和含水率,因而其热值较低,仅为16~19 MJ·kg-1[14]。而生物质液化油含氧量和含水率远低于生物质快速热解油,因而其热值也更高,达到34 MJ·kg-1。生物质快速热解油呈酸性,pH值为2.5,可溶于水,而生物质液化油不溶于水。
生物质油主要是由酸、醛、醇、酯、酮、糖、苯酚、邻甲苯酚、丁香醇、呋喃、木质素衍生取代酚、提取物衍生萜和水等组成的混合物,其组分很复杂,化合物种类多达数百种[15]。这些化合物来源于组成生物质的三大素(纤维素、半纤维素和木质素)的解聚和裂解反应。其中糖类、多种含氧化合物以及呋喃等化合物来自纤维素和半纤维素的裂解,愈创木酚及丁香酚等来自木质素的裂解。
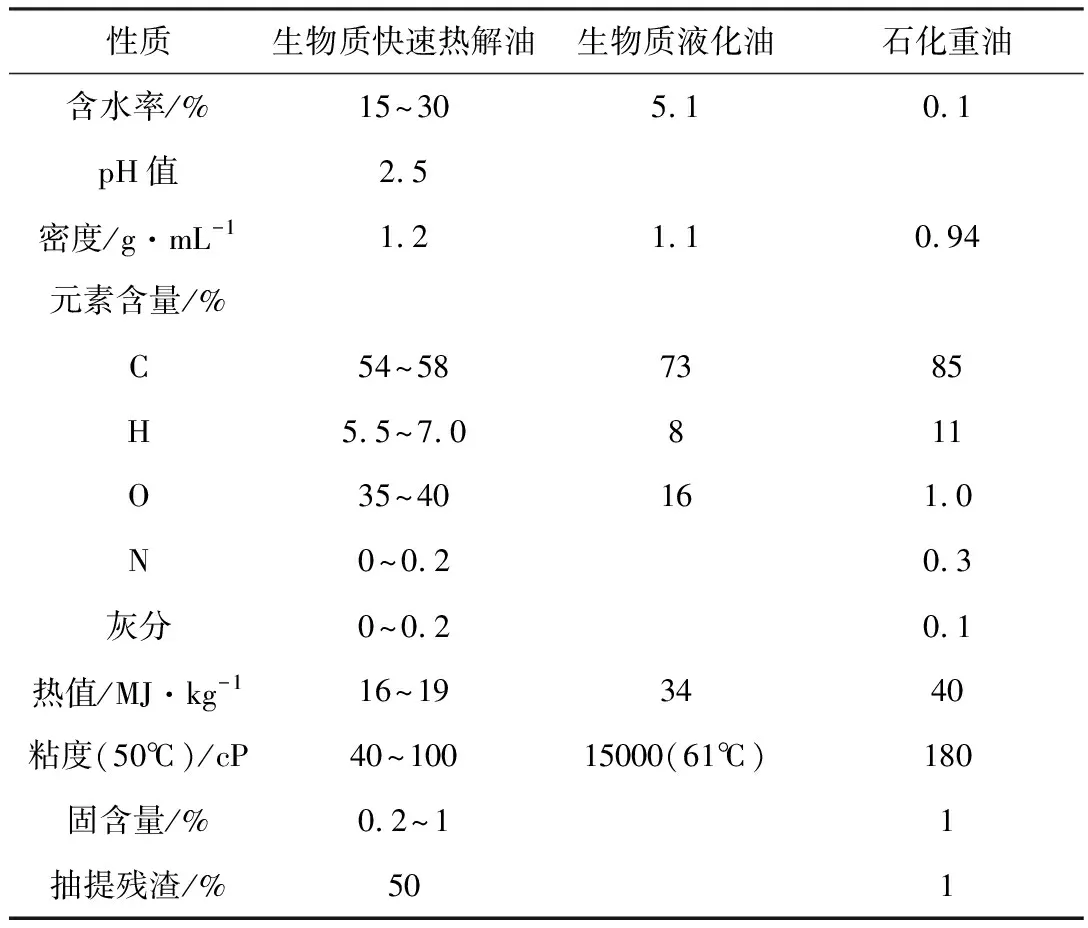
表2 生物质快速热解油、生物质液化油及石化重油的典型性质
Suat等[16]快速热解石榴籽所得油品,经气相色谱-质谱联用仪分析,发现其主要含有苯衍生物、芳香化合物以及酸酯烷烃类多环芳香烃等有机化合物。同时该热解油品中含有大量醛类和酮类化合物,使得其具有很强的亲水性,因此含水率高且水分不易除去。
Sipilä等[17]对稻草和松木等热解制取的生物质油进行分析,并将其分为溶于水的组分(水相)和不溶于水的组分(油相)两大类,经定量测定水相主要成分的组成,发现水相占生物质油质量的60%~80%,主要由水、小分子有机酸和小分子醇组成。油相经正庚烷萃取、柱层析分离后分析发现,甲基呋喃占正庚烷萃取物质量的14.17%、苯乙醇占12.38%、酚类占51%。
Zhang等[18]以乙二醇作溶剂、硫酸作催化剂,在190℃、常压条件下对甘蔗渣进行液化。反应粗产物分离为三个组分:水相组分、丙酮可溶组分以及残渣。通过红外光谱、凝胶渗透色谱和元素分析,发现残渣主要是不溶于丙酮和水的纤维素和木质素衍生物,丙酮可溶物主要是木质素降解物,而水相主要是小分子的醛、酮、醇、酯、酚类以及羧酸类有机化合物。
3 生物质油精制
生物质油由于高含水量、高含氧量、强腐蚀性、强酸性和化学组成复杂等,必须经过精制以降低其含氧量、提高品质。目前生物质油精制方法主要有催化加氢法、催化裂解法、催化酯化法以及分级精制法等。
3.1 催化加氢法
基于石油化工领域广泛应用的催化加氢工艺可以进行生物质油催化加氢。由于生物质油中含氧量远高于硫和氮的量,因而生物质油加氢过程主要是催化加氢脱氧(HDO)。由于生物质油包含大量的不饱和化合物,催化加氢也是对其中的不饱和化合物进行催化加氢反应使其饱和。催化加氢工艺一般在300~400℃、氢气压力7~14 MPa下进行的,催化剂为预硫化处理的CoMo、NiMo、NiW以及Pd/C和Pt/C等[19,20]。生物质油含有大量的酚、醛、酮类物质,因此脱氧主要通过与氢气反应造成碳氧键断裂,使氧元素以H2O或CO2的形式除去。适度加氢可以使反应性较强的不饱和化合物转变为饱和化合物,或使不稳定的醛基转化,从而提高生物质油的稳定性及其能量密度。
Xu等[21]研究镍基催化剂对乙酸的催化加氢,发现Mo-10Ni/γ-Al2O3对乙酸加氢反应活性最大,乙酸加氢后的转化率为33.2%。而将Mo-10Ni/γ-Al2O3用于生物质油的催化加氢可使生物质油pH值从2.16上升到2.84,含水率从46.2%上升到58.99%,H元素含量从6.61%上升到6.93%,同时生物质油的粘度也有一定程度的下降。通过GC-MS分析,精制后的生物质油酯含量比精制前提高了3倍。
Zhang等[22]研究了Co-Mo-P催化加氢裂解生物质快速热解产物中油相组分的反应,发现在反应温度为360℃、反应时间为30 min、氢气压力为2 MPa的条件下,精制生物质油的含氧量从精制前的41.8%降到3%,密度从1.12 g·mL-1降到0.93 g·mL-1。生物质油含氧量的下降,使其热值上升,而密度的降低也使其粘度以及流动性能有所改善,便于运输和储藏。
由于生物质油组成复杂,许多研究均利用与生物质油中主要化合物结构相似的模型化合物来研究生物质油的加氢机理。
Zhao等[23]通过对苯酚以及一些含甲氧基的酚类化合物进行催化加氢,分别以Pd/C、Pt/C、Ru/C、Rh/C为催化剂,在温度为200℃、5 MPa氢气压力下反应30 min,最后模型化合物都能转化成环己醇类化合物,在相同的温度压力条件下加入磷酸溶液,环己醇最后均能转化为环己烷。
Yang等[24]也是以苯酚为模型化合物,研究其催化加氢反应机理。
由于热解生物质油中含有大量的羧酸类物质,因而以乙酸作模型化合物可以研究生物质油中的羧酸类物质以及含羰基化合物的催化加氢反应,并将最优工艺条件直接应用于生物质油的催化加氢反应。
虽然催化加氢技术能大幅降低生物质油含氧量、提高其热值,但是该过程需要消耗大量氢气、使用昂贵的催化剂,且操作条件苛刻,极大地限制了催化加氢技术的推广,目前仍然处于实验室研究阶段。降低氢气的使用量、寻找合适的催化剂、提升催化反应效果、解决催化反应过程中催化剂的结焦失活问题是催化加氢工业化的关键。
3.2 催化裂解法
催化裂解是生物质油在催化剂作用下,将其中的高分子物质裂解成小分子,同时脱除含氧基团。催化裂解反应很复杂,但主要是催化裂解生物质油中的C-C键和C-O键,使高分子裂解,同时使生物质油中的氧以H2O、CO或CO2的形式除去,从而降低生物质油的粘度和含氧量。
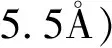

Adjaye等[28]在固定床反应器内采用HZSM-5、H-Y、SiO2、SiO2/Al2O3和沸石对生物质油进行催化裂解。研究发现,利用HZSM-5和沸石作催化剂,芳烃得率大于脂肪烃得率,而利用H-Y、SiO2、SiO2/Al2O3作催化剂获得产物的分布正好相反。同时发现,HZSM-5、H-Y和沸石对生物质油裂解的效能强于其它两种,这说明催化剂酸性越强越有利于生物质油的裂解转化。
催化裂解精制生物质油的产率较低、结焦率高、催化剂寿命短,但反应过程中不需要氢气,反应条件温和,反应设备要求没有催化加氢严格,因而具有很大的发展潜力。由于生物质油的热不稳定性,在温度较高的条件下会发生聚合反应,因此应选择能在较低温度下对生物质油进行高效催化裂解的催化剂。同时也可以在添加供氢溶剂的条件下对生物质油进行催化裂解,尽可能降低反应过程中催化剂的结焦失活。
3.3 催化酯化法
催化酯化是在生物质油中加入醇类助剂,在催化剂的作用下发生酯化等反应,从而将生物质油中的羧基等基团转化为酯类物质。由于羧酸的酯化,生物质油的pH值提高,腐蚀性下降。
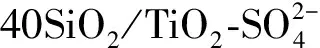
Xiong等[30]在室温条件下利用C6(mim)2-HSO4作催化剂对生物质油进行催化酯化,反应结束后反应产物分为两层,精制油得率为49%。研究发现,催化酯化后的生物质油性能有很大提高,其热值上升到24.6 MJ·kg-1,pH值由2.9上升到5.1、含水率由29.8%降低到8.2%,同时催化剂没有结焦和失活的情况发生。
催化酯化并不能有效降低生物质油的含氧量,但是通过将生物质油中的羧酸转化为酯类,可以增大生物质油的pH值,改善其流动性,提高热值。由于生物质油催化酯化需要添加醇类物质,增加了生物质油精制的成本。
3.4 分级精制法
分级精制是将生物质油进行简单的分离,根据不同组分的特点进行高值化处理。如利用Zou等[31]以及Connors等[32]提出的办法用丙酮抽提生物质油,分离出丙酮不溶残渣,将丙酮可溶组分中的丙酮蒸出后再用正己烷溶解,正己烷可溶物为轻质组分,正己烷不溶物为重质组分,这样就将原生物质油分离成重质组分和轻质组分两部分,其中轻质组分可以应用分级多层利用的理念,首先提取其中集中度高的化学品,然后提取馏程适宜的液体燃料,再利用高沸点馏分中富含的苯丙烷活性组分制备绿色高分子材料;重质组分进行必要的催化裂解处理及精制生产生物质燃料,这样就充分利用了生物质油的各个组分,使生物质油的价值最大化。
分级精制目前应用还不广泛(主要应用于生物质液化油中),但该技术使生物质油的处理更加精细化,对生物质油的精制更有针对性,操作性和实用性更强,因而具有很大的发展潜力。
4 结语
生物质油便于运输和储存,可以分离生产很多高附加值化学品,同时也能部分替代燃料油用于重油锅炉燃烧,经精制后也可以用于柴油发动机和燃气轮机。但目前生物质油精制技术还很不完善,还需要对生物质油的精制机理进行深入研究,寻找合适的反应条件、反应工艺和反应设备,借鉴石油精制原理,为生物质油的精制过程寻找新的思路。通过开发性能稳定的催化剂,破解催化剂结焦失活难题。另外,针对生物质油含有的物质以及可能发生的化学反应,尝试将生物质油中有价值的成分在简单装置上通过合适的反应转化为高附加值的产品,也是高效利用生物质油的一个突破口。
参考文献:
[1] 朱锡锋,陆强.生物质快速热解制备生物油[J].科技导报,2007,25(21):69-75.
[2] Behrendt F,Neubauer Y,Oevermann M,et al.Direct liquefaction of biomass[J].Chemical Engineering & Technology,2008,31(5):667-677.
[3] 陆强,朱锡锋,李全新,等.生物质快速热解制备液体燃料[J].化学进展,2007,19(8):1064-1071.
[4] Balat M.Mechanisms of thermochemical biomass conversion pro-cesses.Part 3:Reactions of liquefaction[J].Energy Sources,Part A:Recovery,Utilization,and Environmental Effects,2008,30(7):649-659.
[5] Balat M.Mechanisms of thermochemical biomass conversion pro-cesses.Part 1:Reactions of pyrolysis[J].Energy Sources,Part A:Recovery,Utilization,and Environmental Effects,2008,30(7):620-635.
[6] Demirbas M F.Biorefineries for biofuel upgrading:A critical review[J].Applied Energy,2009,86(1):S151-S161.
[7] Mohan D,Pittman C U,Steele P H,et al.Pyrolysis of wood/biomass for bio-oil:A critical review[J].Energy & Fuels,2006,20(3):848-889.
[8] Jegannathan K R,Chan E S,Ravindra P,et al.Harnessing biofuels:A global renaissance in energy production?[J].Renewable and Sustainable Energy Reviews,2009,13(8):2163-2168.
[9] Huber G W,Dumesic J.An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery[J].Catalysis Today,2006,111(1-2):119-132.
[10] 姚向君,田宜水.生物质能资源清洁转化利用技术[M].北京:化学工业出版社,2006:120.
[11] 邹献武,杨智,秦特夫.木材正辛醇液化产物的红外光谱分析[J].光谱学与光谱分析,2008,29(6):1545-1548.
[12] Chumpoo J,Prasassarakich P.Bio-oil from hydro-liquefaction of bagasse in supercritical ethanol[J].Energy & Fuels,2010,24(3):2071-2077.
[13] Czernik S,Bridgwater A V.Overview of applications of biomass fast pyrolysis oil[J].Energy & Fuels,2004,18(2):590-598.
[14] Lu Q,Wen Z H,Zhu X F.Overview of fuel properties of biomass fast pyrolysis oils[J].Energy Conversion and Management,2009,50(5):1376-1383.
[15] Branca C,Giudicianni P,Colomba B.GC/MS Characterization of liquids generated from low-temperature pyrolysis of wood[J].Industrial & Engineering Chemistry Research,2003,42(14):3190-3202.
[16] Suat Uçar,Selhan Karagöz.The slow pyrolysis of pomegranate seeds:The effect of temperature on the product yields and bio-oil properties[J].Journal of Analytical and Applied Pyrolysis,2009,84(2):151-156.
[17] Sipilä Kai,Kuoppala Eeva,Fagernäs Leena,et al.Characterization of biomass-based flash pyrolysis oils[J].Biomass and Bioenergy,1998,14(2):103-113.
[18] Zhang T,Zhou Y J,Liu D H.Qualitative analysis of products formed during the acid catalyzed liquefaction of bagasse in ethylene glycol[J].Bioresource Technology,2007,98(7):1454-1459.
[19] Elliott D C.Historical development in hydroprocessing bio-oil[J].Energy & Fuels,2007,21(3):1972-1815.
[20] Elliott D C,Beckman D,Bridgwater A V,et al.Developments in direct thermochemical liquefaction of biomass:1983-1990[J].Energy & Fuels,1991,5(3):399-410.
[21] Xu Y,Wang T,Ma L L,et al.Upgrading of liquid fuel from the vacuum pyrolysis of biomass over the Mo-Ni/gamma-Al2O3catalysts[J].Biomass and Bioenergy,2009,33(8):1030-1036.
[22] Zhang S,Yan Y,Li T,et al.Upgrading of liquid fuel from the pyrolysis of biomass[J].Bioresource Technology,2005,96(5):545-550.
[23] Zhao C,Kou Y,Lemonidou A A,et al.Highly selective catalytic conversion of phenolic bio-oil to alkanes[J].Angewandte Chmie,2009,48(22):3987-3990.
[24] Yang Yun,Allan Gilbert,Xu C B.Hydrodeoxygenation of bio-crude in supercritical hexane with sulfided CoMo and CoMoP catalysts supported on MgO:A model compound study using phenol[J].Applied Catalysis A:General,2009,360(2):242-249.
[25] Peng J,Chen P,Lou H,et al.Catalytic upgrading of bio-oil by HZSM-5 in sub-and super-critical ethanol[J].Bioresource Technology,2009,100(13):3415-3418.
[26] 郭晓亚,颜涌捷,任铮伟.生物质油精制中催化剂的应用及进展[J].太阳能学报,2003,24(2):206-212.
[27] Vitolo S,Seggiani M,Frediani P,et al.Catalytic upgrading of pyrolytic oils to fuel over different zeolites[J].Fuel,1999,78(10):1147-1159.
[28] Adjaye J D,Katikaneni S,Bakhshi N.Catalytic conversion of a biofuel to hydrocarbons:Effect of mixtures of HZSM-5 and silica-alumina catalysts on product distribution[J].Fuel Processing Technology,1996,48(2):115-143.
[29] Zhang Q,Chang J,Wang T J,et al.Upgrading bio-oil over different solid catalysts[J].Energy & Fuels,2006,20(6):2717-2720.
[30] Xiong W M,Zhu M Z,Deng L,et al.Esterification of organic acid in bio-oil using acidic ionic liquid catalysts[J].Energy & Fuels,2009,23(4):2278-2283.
[31] Zou X W,Qin T F,Yang Z,et al.Mechanisms and main regularities of biomass liquefaction with alcoholic solvents[J].Energy & Fuels,2009,23(10):5213-5218.
[32] Connors W J,Johanson L N,Sarkanen K V,et al.Thermal degradation of Kraft lignin in tetralin[J].Holzforschung,2009,34(1):29-37.
