脑外伤大鼠应激性溃疡与内毒素血症和脂质过氧化的相关性研究
2010-03-17王天懿孟庆颖朱玉群杨昭徐
王天懿,孟庆颖,朱玉群,杨昭徐
(首都医科大学附属北京天坛医院消化内科,北京 100050)
创伤性脑损伤(traumatic brain injury,TBI)并发应激性溃疡(stress ulcer,SU)的相关研究较多[1-2],认为缺血、再灌注损伤可引起脂质过氧化(lipid peroxidation,LPO)反应,但发病机制仍未完全阐明。近年研究发现TBI并发肝损伤也不少见[3],肝损伤后可引起内毒素血症(endotoxemia,ET),激发全身炎症反应综合症(systemic inflamatory response syndrome,SIRS),对机体造成再次打击,进一步诱发多器官功能不全综合症(multiple organ dysfunction syndrome,MODS)。本研究拟通过观察大鼠TBI后胃粘膜和肝损伤,结合血清肝酶、MDA和SOD、血浆脂多糖(lipopolysaccharide,LPS)的变化,探讨TBI后SU与ET和LPO的关系。
1 材料与方法
1.1 实验动物及分组
32只雄性Wister大鼠,体质量250 g~280 g,随机分4组:正常对照组(C)、TBI后6、12、24 h组(T6、T12、T24),每组8只。改良Feeney自由落体撞击法[4]建立TBI并发SU模型。
1.2 实验方法
1.2.1 溃疡指数(UI) 依Guth标准[5]评定。
1.2.2 病理学检查 取胃大弯侧前壁组织和肝右后叶组织标本,光镜、透射及扫描电镜观察组织学改变。
1.2.3 血清ALT、AST测定 左心室取血,酶法检测ALT、AST。
1.2.4 血浆LPS测定 门静脉取血,鲎试验试剂盒及LPS标准品测定。试剂盒由上海伊华临床医学科技公司提供。
1.2.5 胃黏膜MDA、SOD测定 刮取胃后壁黏膜制备组织匀浆,硫代巴比妥酸法测定MDA,化学发光法测定SOD,Lorry法测定匀浆液蛋白含量。试剂盒由南京生物工程研究所提供。
1.3 统计学分析
2 结果
2.1 大鼠TBI对溃疡指数的影响
TBI后6 h胃黏膜轻度充血、水肿,少量点状糜烂;12 h见明显充血,点线状糜烂和新鲜出血灶;24 h见点线状糜烂、剥脱、小溃疡形成;TBI各组UI与对照组比较有统计学意义(P<0.05)。结果见表1。
大鼠TBI的血清ALT、AST变化 TBI后6 h血清ALT开始显著升高,伤后12 h血清AST开始显著升高。见表1。
大鼠TBI对血浆LPS变化 TBI各组与对照组比较,LPS水平均显著升高,见表1。
表1 血清ALT、AST及门静脉LPS水平变化
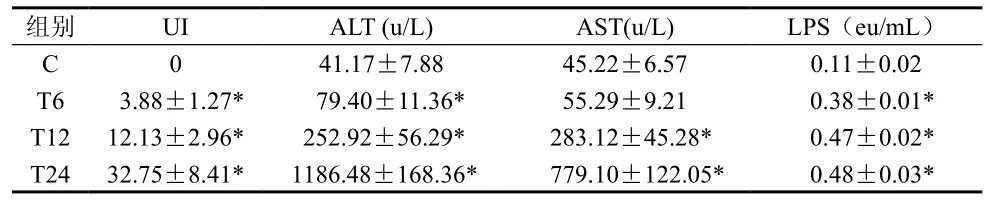
表1 血清ALT、AST及门静脉LPS水平变化
注:与C组比较*P<0.05。
组别 UI ALT (u/L) AST(u/L) LPS(eu/mL)C 0 41.17±7.88 45.22±6.57 0.11±0.02 T6 3.88±1.27* 79.40±11.36* 55.29±9.21 0.38±0.01* T12 12.13±2.96* 252.92±56.29* 283.12±45.28* 0.47±0.02* T24 32.75±8.41* 1186.48±168.36* 779.10±122.05* 0.48±0.03*
2.2 大鼠TBI对胃黏膜病理学改变
光镜显示:TBI后6 h黏膜脱失明显,黏膜内水肿,毛细血管充血,中性粒细胞浸润;12 h及24 h黏膜出血、坏死、脱落,形成溃疡(见图1~2)。扫描电镜:TBI后6 h胃黏膜皱褶欠规整,胃小凹消失,细胞大小不一,可见破损;12 h胃小凹消失,细胞破损、缺失,形成空洞;24 h可见微绒毛消失,较多细胞破损、缺失,裸露(见图3~4)。
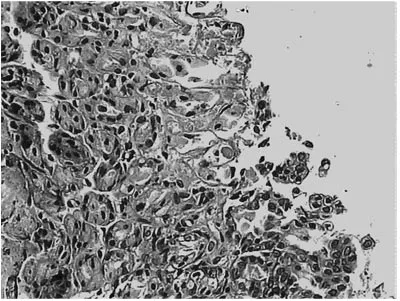
图1 大鼠TBI 6 h胃黏膜脱失明显,黏膜内水肿,毛细血管充血,中性粒细胞浸润光镜图(HE×200)

图2 大鼠TBI 24 h黏膜出血、坏死、脱落,形成溃疡,局限于黏膜层光镜图(HE×100)

图3 大鼠TBI 12 h胃小凹消失,细胞破损、缺失,形成空洞扫描电镜图(×3000)
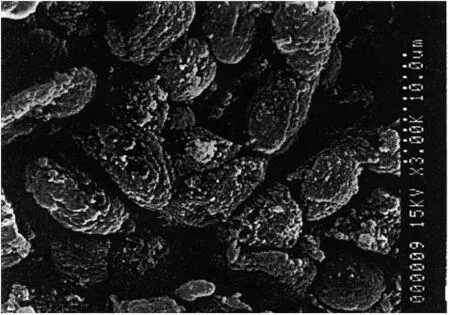
图4 大鼠TBI 24 h 微绒毛消失,黏液消失,大部分细胞破损、缺失,裸露于胃腔表面扫描电镜图(×3000)
2.3 大鼠TBI对肝组织病理学改变
光镜显示:TBI后6 h可见肝组织轻度充血、水肿;12 h可见细胞肿胀,空泡变性;24 h见胞浆广泛水肿、气球样变,少许核破碎和灶性坏死(见图5~6)。透射电镜显示:TBI后6 h见肝窦扩张、Disse间隙增宽,少量核染色质边集;12 h见较多空泡变,广泛核染色质凝聚边集;24 h见广泛胞浆水肿,大量空泡形成,枯否细胞增多,粗面和滑面内质扩张、脱颗粒,核破碎、融合(见图7~8)。
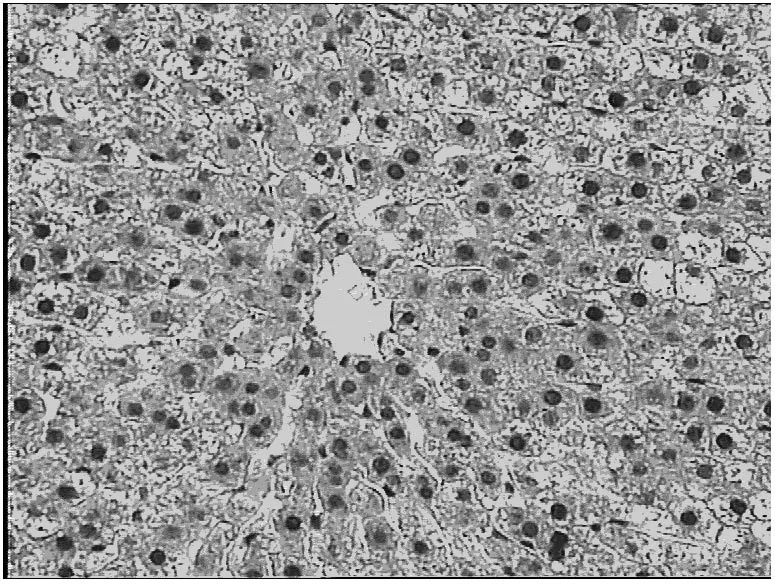
图5 大鼠TBI 12 h肝细胞肿胀,细胞浆疏松浅染,部分细胞空泡变性光镜图(HE×200)
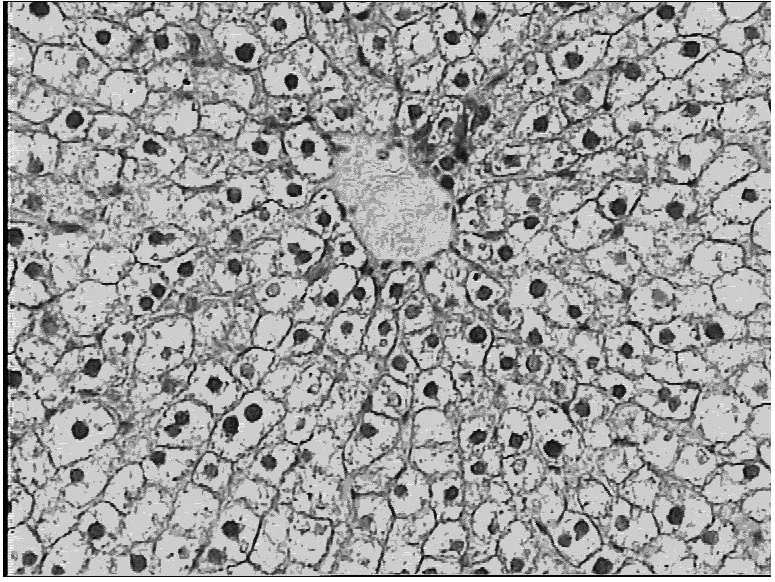
图6 大鼠TBI 24 h肝细胞结构不清,细胞普遍增大,胞浆广泛水肿、气球样变,核破碎,灶性坏死光镜图(HE×200)
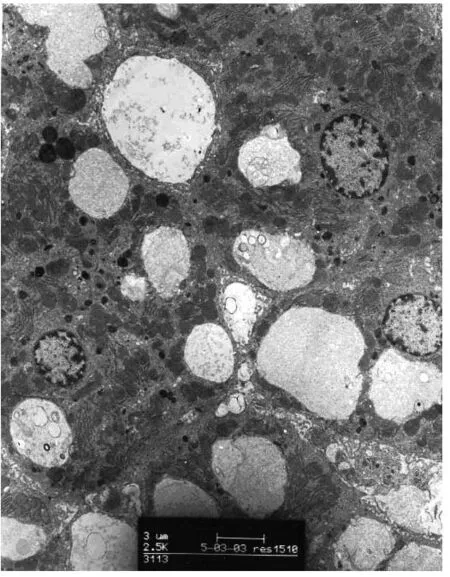
图7 大鼠TBI 12 h后可见多而大的空泡样物透射电镜图(×25000)
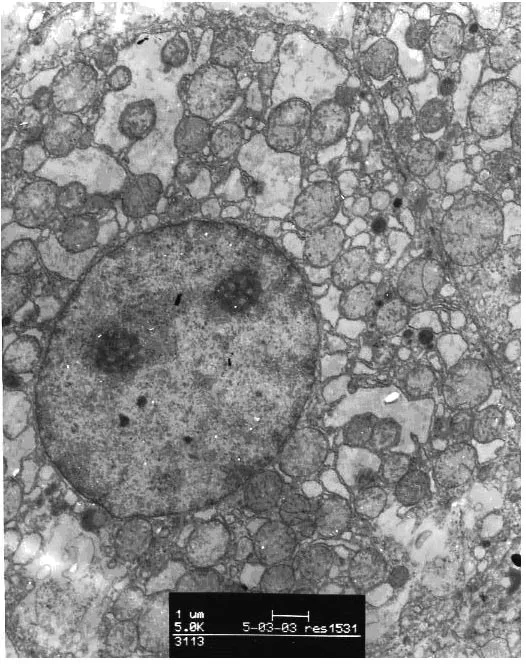
图8 大鼠TBI 24 h后线粒体增多、肿胀,内质网高度扩张透射电镜图(×5000)
2.4 大鼠TBI对胃黏膜MDA、SOD变化
TBI后胃黏膜MDA显著升高,SOD显著降低。结果见表2。
表2 大鼠TBI对胃黏膜组织MDA和SOD变化

表2 大鼠TBI对胃黏膜组织MDA和SOD变化
注:与C组比较*P<0.05。
组别 MDA nmol/mg.pro. SOD u/mg.pro. C 3.12±0.30 137.03±5.02 T6 13.06±1.82* 75.41±7.55* T12 14.34±3.01* 62.71±5.96* T24 14.52±2.08* 61.20±5.73*
2.5 血浆LPS与肝酶、胃黏膜MDA、SOD相关性分析
TBI后各组血浆LPS与ALT、AST和胃黏膜组织MDA呈显著正相关(r分别为0.77、0.79和0.75,P<0.05),与SOD呈显著负相关(r分别为-0.69,P<0.05)。
3 讨论
SU是TBI后常见并发症,一旦出现消化道出血,不仅增加治疗的复杂性,也是病情加重甚至导致死亡的重要原因。文献报道重度TBI并发消化道出血的死亡率达40%~80%[6]。因此,有效防治SU对提高TBI患者生存率至关重要。本研究中大鼠TBI后6 h即有胃黏膜细胞的水肿、变性,12 h、24 h出现上皮细胞破损、脱落、溃疡形成,支持TBI早期即可发生胃黏膜损伤的观点。
目前关于TBI后SU的发生机制仍不十分清楚,多认为与神经、体液、激素水平和局部细胞内环境发生变化有关。一方面,交感神经强烈兴奋,胃黏膜血管强烈收缩,组织缺血、缺氧直接损害胃粘膜[7]。另一方面,缺血再灌注损伤引起黏膜组织LPO反应,LPO使氧自由基(OFRs)的产生与清除失衡,活性氧在体内堆积引起细胞毒性。自由基清除剂SOD、LPO代谢产物MDA是公认的能较好反映体内LPO水平的间接指标。本研究中,TBI后胃黏膜MDA显著升高,SOD明显降低,与病理变化一致,提示LPO可能是TBI后SU的重要致病因素。LPO产生大量OFRs,OFRs对细胞膜有直接毒性作用,能与多种细胞成分(膜磷脂、蛋白、核酸等)发生反应,影响膜通透性及离子转运,干扰线粒体氧化磷酸化,使能量产生障碍;同时,OFRs引起中性蛋白酶活性的病理性增加,使细胞膜结构分解,导致细胞死亡[8]。
TBI后内脏的缺血再灌注损伤,也可直接损害肝细胞[9]。本研究中大鼠TBI后血清ALT、AST明显升高,肝组织超微结构也显示不同程度的病理变化,提示肝脏也是TBI后早期易受损伤的脏器之一。
由于肝脏的特殊功能,肝损伤发生后,可导致细菌和内毒素“逃逸”,引起菌血症和ET[10]。本研究中TBI后大鼠血浆LPS水平明显升高,与肝组织和胃黏膜的病理改变一致,且与血清肝酶及胃黏膜MDA、SOD变化显著相关,表明肝损伤和ET可能参与或促进了TBI后SU的发生发展。肝脏库普弗细胞被LPS大量激活后,可通过合成和分泌多种炎性介质,进一步激发组织的LPO反应[11],对机体产生再次或多次打击,引发“瀑布效应”,造成原发灶和远隔器官的损伤,最终可出现MODS[12]。ET还进一步加重黏膜屏障的微循环障碍,在炎症细胞因子的参与下更加重胃黏膜损伤,最终导致胃黏膜糜烂、出血和溃疡形成。
因此,我们认为,TBI后ET和LPO通过级联或协同作用,引起或加重SU和肝损伤。
[1] 肖正武,邵忠华.重型颅脑损伤致应激性溃疡48例的临床分析[J].浙江创伤外科,2009,02:143.
[2] 袁 平.颅脑外伤后并发急性胃黏膜病变64例临床分析[J].贵州医药, 2009,01:34-35.
[3] 孟庆颖,朱玉群.成人TBI后早期应激性肝损害的临床分析[J].首都医科大学学报,2007,28(4):513-515.
[4] Feeney DM,Boyeson MG,Linn RT,et al.Response to cortical injury:I Methodology and local effects of contusions in the rat[J].Brain Res, 1981,211(1):67-77.
[5] Guth PH,Aure D,Paulsen G..Topical aspirin plus HCL gastric lesion in the rat.Cytoprotective effect of prostagiandin,Cimetidine and probanthine[J].Gastroenterology, 1979,76:88.
[6] 高英丽,朱京慈.颅脑损伤后应激性溃疡的发病机制及预防[J].中华创伤杂志,2005,06:478-479.
[7] 杜秀芳,杨 拯,孟 玲,等.应激性胃溃疡的治疗研究进展[J].现代预防医学.2008,19:(3)858-859.
[8] Tamagno E,Parola M,Guglielmotto M,et al.Multiple signaling events in amyloidβ-induced,oxidative stress-dependent neuronal apoptosis[J].Free Radic Biol Med,2003,35(1):45.
[9] Tselepidis S,Papazoglou L,Dessiris A,et al.Liver injury after ischemia and reperfusion:the role of oxygen free radicals[J].Mil Med,2004,169(7):531-535.
[10] 王春妍,杨世忠,江海艳.急性肝损伤大鼠肠源性内毒素血症形成机理及其作用的实验研究[J].临床肝胆病杂志,2007,23(2):109-111.
[11] Ding H,Peng R,Reed E,et al.Effects of Kupffer cell inhibition on liver function and hepatocellular activity in mice[J].Int J Mol Med.2003,12(4):549-557.
[12] Kinoshita M,Uchida T,Nakashima H,et al.Opposite effects of enhanced tumor necrosis factor-alpha production from Kupffer cells by gadolinium chloride on liver injury/mortality in endotoxemia of normal and partially hepatectomized mice[J].Shock,2005,23(1):65-72.
