超高效液相色谱-串联质谱法测定大豆分离蛋白中的嘌呤
2024-11-05彭静雅林钦恒潘云山张飞蔡杏宜宋思园黄志峰




摘要 [目的]建立同时测定大豆分离蛋白中4种嘌呤的超高效液相色谱-串联质谱(UPLC-MS/MS)方法。[方法]采用三氟乙酸∶甲酸(V∶V=1∶1)将样品中的嘌呤进行水解和提取,并用水稀释。以含0.002%甲酸水溶液-甲醇体系为流动相,选择安捷伦超高压色谱柱 Poroshell 120 EC-C18分离4种嘌呤化合物(鸟嘌呤、腺嘌呤、次黄嘌呤和黄嘌呤),以质谱条件为电喷雾(ESI)正离子电离,MRM模式,外标法定量测定。[结果]在1~200 ng/mL浓度范围内,4种嘌呤均具有较好的线性关系,相关系数(r)均不低于0.999 定量限(LOQ)和检出限(LOD)分别在0.24~3.00和0.08~1.00 mg/kg。在3个浓度级别的加标水平下,4种嘌呤的平均回收率在81.7%~95.2%,相对标准偏差(RSD)均低于8.6%(n=6)。[结论]该方法可操作性强、简捷快速,具有较高的灵敏度、回收率和精密度,可以同时检测大豆分离蛋白中的4种嘌呤化合物。
关键词 超高效液相色谱-串联质谱法;嘌呤;大豆分离蛋白
中图分类号 TS 207 文献标识码 A
文章编号 0517-6611(2024)20-0179-04
doi:10.3969/j.issn.0517-6611.2024.20.043
开放科学(资源服务)标识码(OSID):
Determination of Purines in Soybean Protein Isolate by Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry
PENG Jing-ya 3,LIN Qin-heng 3,PAN Yun-shan 3 et al
(1.Institute of Analysis,Guangdong Academy of Sciences (China National Analytical Center,Guangzhou),Guangzhou,Guangdong 510070;2.Guangdong Provincial Key Laboratory of Chemical Measurement and Emergency Test Technology,Guangzhou,Guangdong 510070;3.Guangdong Provincial Engineering Research Center for Efficacy Component Testing and Risk Substance Rapid Screening of Health Food,Guangzhou,Guangdong 510070)
Abstract [Obiective]To establish a method for the simultaneous measurement of 4 purines in soybean protein isolate (SPI) using UPLC-MS/MS.[Method]Purines in SPI samples were hydrolyzed and extracted with trifluoroacetic acid:formic acid (V∶V=1∶1),and diluted with water.The 4 purines (guanine,adenine,hypoxanthine and xanthine) were then separated on an Agilent Poroshell 120 EC-C18 column,using 0.002% formic acid aqueous solution and methanol as mobile phases with gradient elution.Subsequently,the target compounds were analyzed with electrospray ionizationsource in positive ion mode under multi-reaction monitoring mode,employing the external standard quantitative method.[Result]Within the concentration range of 1-200 ng/mL,all four purines had good linear relationships,with correlation coefficients (r) exceeding 0.999 1.The limits of quantitation (LOQ) and the limits of detection (LOD) for the four purines were in the range of 0.24-3.00 mg/kg and 0.08-1.00 mg/kg.The spiked experiments were performed at three concentration levels,and the average recovery rates for the four components ranged from 81.7% to 95.2%,with RSD all less than 8.6%(n=6).[Conclusion]This method is highly operable,simple and fast,with high sensitivity,recovery rate and precision,and can simultaneously detect four purine compounds in soy protein isolate.
Key words Ultra performance liquid chromatography-tandem mass spectrometry;Purine;Soybean protein isolate
大豆是一种优良的粮食作物,不仅营养丰富、口味佳,还兼具保健功能。大豆的蛋白质含量约占其总干重的40%[1],是食物中非常重要的蛋白质来源,相关的深加工产品也已得到充分的开发,如大豆粉、分离蛋白、浓缩蛋白和组织蛋白等[2]。其中大豆分离蛋白(soybean protein isolate,SPI)是一种全价蛋白制品,以低温脱溶大豆粕为原料,经过碱溶酸沉法等工艺生产而得[3]。SPI的外观为乳白色至淡黄色粉末,一般含有多种氨基酸成分和不低于90%的蛋白质含量[4],是一种优良的蛋白质原料。SPI还表现出良好的食品加工性能,被广泛应用于乳制品、肉制品、面制品和抗菌保鲜包装等领域[5-6],同时因其具有保健作用,在保健食品和医药领域也有一定的应用[7]。
嘌呤类物质是生物体内核酸的主要组成部分,包含鸟嘌呤、腺嘌呤、次黄嘌呤、黄嘌呤和各自的衍生物,具有多种生物学功能,对机体的能量供应和新陈代谢起着关键作用[8-9]。如果机体内嘌呤物质的代谢长期出现紊乱,将会发展成高尿酸血症,并极有可能导致痛风等代谢性疾病[10]。研究表明,限制饮食中嘌呤的摄入可预防高尿酸血症或减轻其相关症状[11]。大豆及其相关制品一直都被认为是高嘌呤食物的代表[12],但有报道称,大豆蛋白中嘌呤含量为0.388 mg/g[13],而SPI在其加工过程中嘌呤脱除率也较高[14],表明这2类大豆制品应属于低嘌呤食物。目前关于SPI嘌呤含量的研究较少,SPI是否为高嘌呤食物仍存在争议,因此有必要开发相关的测试方法,更深入地研究SPI的嘌呤含量,才能更科学地将其应用于膳食食品中。
目前鲜见SPI中嘌呤测定方法的相关报道,食品中常见的嘌呤测试方法主要有毛细管电泳色谱法[15]、高效液相色谱法[16]和液相色谱-串联质谱法[17]等,后2种是目前较主流的检测方法。嘌呤为弱碱性化合物,在反向高效液相色谱中保留较弱、分离度较差,导致干扰因素多、准确率低,需要结合三重四极杆串联质谱法测定才能提高结果的准确性。该试验在相关研究的基础上,采用三氟乙酸与甲酸的混合酸对SPI样品中的嘌呤组分进行水解和提取,并结合超高效液相色谱-串联质谱法,建立了同时检测SPI中4种嘌呤(鸟嘌呤、腺嘌呤、次黄嘌呤、黄嘌呤)的方法,以期为充分了解SPI中的嘌呤含量提供测试手段和参考依据。
1 材料与方法
1.1 试材与试剂
标准品:鸟嘌呤、腺嘌呤、黄嘌呤(含量≥99.8%),采购自上海诗丹德标准技术服务有限公司,次黄嘌呤(含量99.6%),采购自美国sigma-aldrich公司;甲醇(色谱纯,美国honeywell公司);甲酸、三氟乙酸(色谱纯,上海麦克林生化科技有限公司);氢氧化钠、盐酸(分析纯,广州化学试剂厂);SPI粉末样品为实验室留样;实验室用水为蒸馏水(广州屈臣氏食品饮料有限公司)。
1.2 仪器与设备
1290型超高效液相色谱仪,配有6460A型三重四极杆质谱仪(美国安捷伦公司);FB15067型超声波清洗器(美国赛默飞公司);HWS26型电热恒温水浴锅(上海恒一科学仪器公司);XW-80A型旋涡振荡器(上海琪特分析仪器公司);BSA224S型电子天平(德国赛多利斯公司,感量0.000 1 g)。
1.3 试验方法
1.3.1 前处理方法。将SPI样品均质后,称取约0.2 g样品(精确至0.001 g)于25 mL试管中,加入1 mL水超声振荡至样品溶解。加入6 mL三氟乙酸∶甲酸(V∶V=1∶1),立即振荡1 min,轻轻盖上试管塞(勿拧紧),并置于90 ℃水浴中水解15 min。水解结束后待其恢复至室温,调节水解液的pH为接近中性,最后用水定容至25 mL并充分摇匀。根据样品的嘌呤含量,用水进一步稀释至合适浓度。样液经 0.22 μm PTFE滤膜过滤后,供上机测定。
1.3.2 标准溶液配制。分别准确称取鸟嘌呤、腺嘌呤、次黄嘌呤和黄嘌呤标准品约25 mg,分别用水超声溶解,可加入少量的氢氧化钠或稀盐酸溶液助溶,定容至25 mL,配制成1.0 mg/mL的单一嘌呤储备溶液。然后分别精密吸取上述4种储备溶液1 mL,混合,并用水定容至100 mL,稀释成10 μg/mL 的混合标准溶液。上述溶液均置于4 ℃保存。上机测定前,将混合标准溶液根据需求,现配现用,进一步稀释成终浓度为1~200 ng/mL的标准曲线工作溶液。
1.3.3 色谱条件。超高压色谱柱Poroshell 120 EC-C18 (美国安捷伦公司,3.0 mm×150 mm,2.7 μm);柱温35 ℃;流动相为含0.002%甲酸的水溶液(A)-甲醇(B),梯度洗脱程序:0~5.5 min,0~5% B;5.5~5.6 min,5%~95% B;5.6~8.0 min,95% B;8.0~8.1 min,95%~0% B;8.1~12.0 min,0% B;流速0.4 mL/min;进样量2 μL。
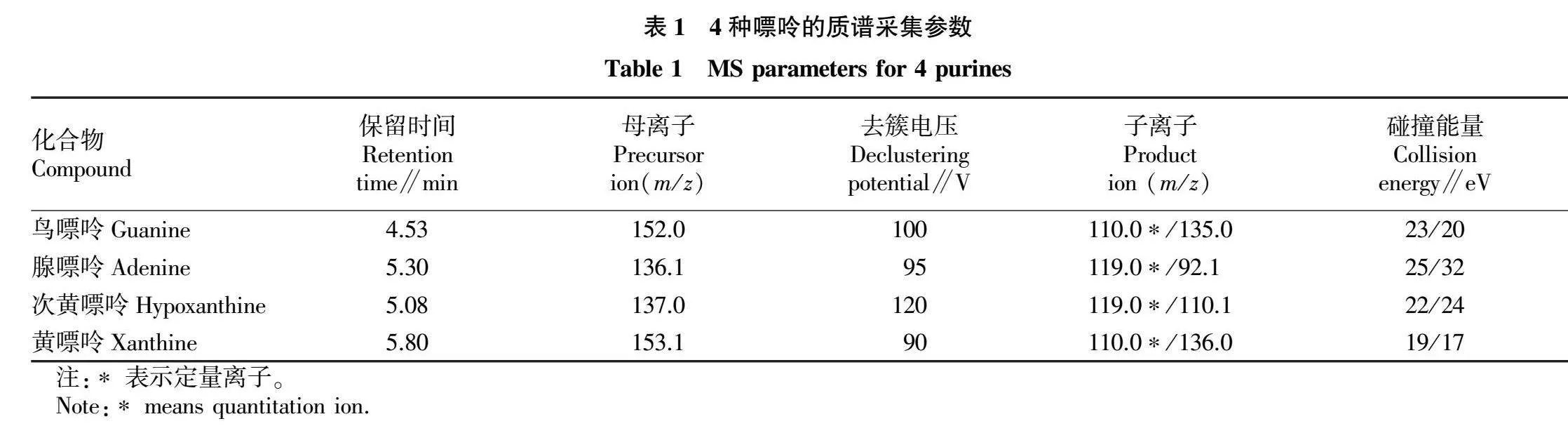
1.3.4 质谱条件。AJS ESI离子源,带有鞘气流;使用正离子扫描方式和多反应离子监测模式(MRM);毛细管电压为3.5 kV;雾化气为310.26 kPa;干燥气和鞘气,流速分别为6.0和10.0 L/min,温度均为350 ℃。4种嘌呤化合物的色谱保留时间和质谱分析参数详见表1。
2 结果与分析
2.1 酸水解条件的优化
嘌呤包括结合态嘌呤和游离态嘌呤,生物体中的嘌呤主要以结合态为主。结合态嘌呤必须水解为游离态才能被测定,水解方式一般采用酸水解,常用的酸类主要是高氯酸或三氟乙酸与甲酸的混合酸。有研究表明,采用三氟乙酸∶甲酸(V∶V=1∶1)混合酸(以下简称TF混合酸)进行水解,嘌呤损失少,效果较好[18-19]。SPI样品中蛋白质含量较高,可以参考高蛋白含量的样品如肉类或蛋白样品等的酸水解条件。杨平等[20]测定猪里脊肉中嘌呤含量采用的酸水解条件为:0.2 g 肉泥加入1 mL超纯水,再加入10 mL TF混合酸,90 ℃水浴水解12 min;武惠敏等[13]测定酵母蛋白中嘌呤含量采用的酸水解条件为:0.200 g样品加入1 mL 水,再加入10 mL TF混合酸,85 ℃水浴水解15 min。故SPI的水解可以考虑采用TF混合酸在90 ℃水浴中水解15 min,但由于SPI为十分细腻的粉末且易溶于水,酸的用量不宜过多。孙玉凤等[21]测定大豆粉末中嘌呤含量采用的酸水解条件为:0.100 g样品加入1 mL 水混匀,再加入1 mL TF混合酸,100 ℃水浴水解30 min,大约相当于0.2 g样品仅加入1 mL TF混合酸水解;同时由于酸的用量减少,其水解的温度和时间都有所提升,而水解时间的延长也降低了前处理的效率。因此,为了同时保证前处理的效果和效率,有必要进行试验来确定 TF 混合酸的适宜用量。
综合参考上述方法,该研究选择以90 ℃水浴水解15 min为固定条件,并以2个不同来源的SPI样品为对象,比较考察了2、4、6、8、10 mL的TF混合酸用量对嘌呤的水解提取效果。前处理按“1.3.1”的方法进行(其中TF混合酸的用量分别加入2、4、6、8、10 mL),再按“1.3.3”和“1.3.4”的条件上机测定,结果见表2。由表2可知,2个SPI样品中,鸟嘌呤和腺嘌呤含量均较高,次黄嘌呤含量较低,而黄嘌呤则未检出;同时,鸟嘌呤、腺嘌呤和总嘌呤的含量在TF混合酸用量为6 mL时均达到了峰值,随着TF混合酸用量继续增加,嘌呤含量反而有所下降,这说明过量的TF混合酸会导致游离态的嘌呤发生降解,使嘌呤含量偏低。故该试验选择TF混合酸的用量为6 mL。
2.2 色谱条件的选择
色谱柱采用安捷伦 Poroshell 120 EC-C18,固定甲醇为有机洗脱液,对3种无机流动相(水、含1 mmol/L 乙酸铵水、含0.002%甲酸水)分离4种嘌呤的效果进行考察。试验发现,不同的无机流动相对腺嘌呤的保留时间影响较大,对另外3种嘌呤的保留时间影响则较小。以水-甲醇为流动相时,腺嘌呤保留时间为6.10 min,与其他3种嘌呤的分离度较好,但鸟嘌呤和次黄嘌呤的出峰时间分别为4.85和4.90 min,两者十分接近;以含1 mmol/L乙酸铵水-甲醇为流动相时,腺嘌呤的保留时间大大提前,但与鸟嘌呤重叠,两者保留时间皆为3.6 min,分离效果不佳;当流动相采用含0.002%甲酸水-甲醇体系时,4种嘌呤化合物的出峰都没有重叠,峰型较好,分离度相对达到最优(图1)。因此,该试验的无机流动相选择含0.002%甲酸的水溶液。
2.3 质谱条件优化
以4种嘌呤化合物浓度为0.5 μg/mL的单一标准溶液为目标物,进行一级质谱全扫描(负离子、正离子检测模式分别进行),结果显示4种嘌呤化合物均在正离子模式下表现出较高的响应值,能形成[M+H]+准分子离子峰。然后在正离子模式下继续优化[M+H]+离子的去簇电压,并利用二级质谱MRM模式确定4种嘌呤化合物的定性定量子离子及其最优碰撞能量,最终得到4种嘌呤化合物在仪器上的最佳参数。
2.4 方法学验证
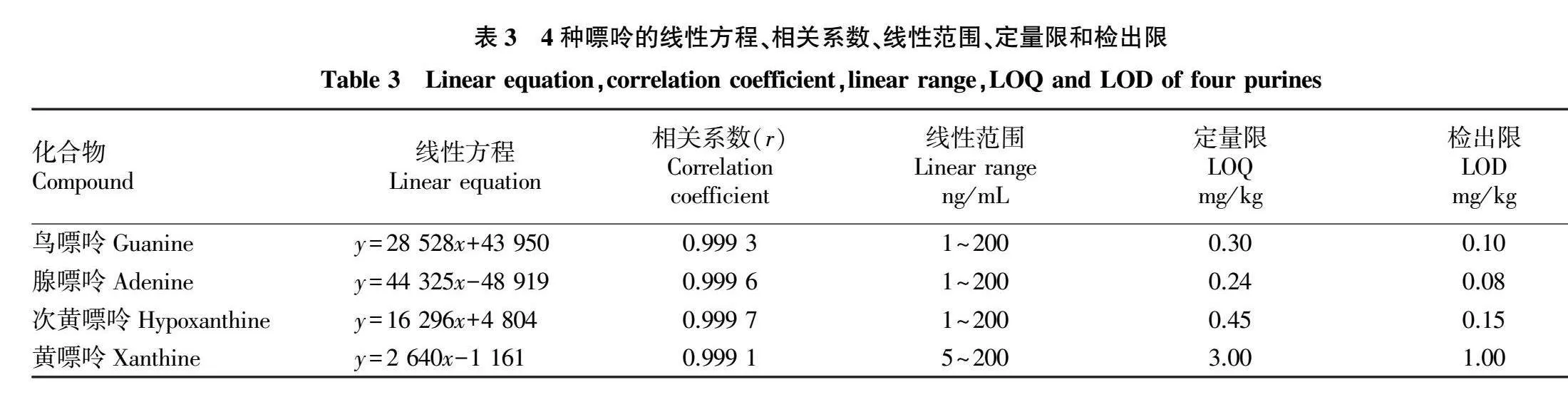
2.4.1 线性关系、定量限及检出限。将“1.3.2”的标准曲线工作溶液按“1.3.3”色谱条件和“1.3.4”质谱条件进行分析,考察4种嘌呤目标物的线性关系。将分析结果以定量离子响应值对目标物质量浓度进行线性回归,计算其线性方程、相关系数和线性范围,定量限(LOQ)和检出限(LOD)则按照各目标物定量离子的10倍信噪比和3倍信噪比所对应的浓度分别计算,结果见表3。鸟嘌呤、腺嘌呤和次黄嘌呤在1~200 ng/mL、黄嘌呤在5~200 ng/mL均表现出较好的线性关系,相关系数(r)在0.999 1~0.999 7,4种嘌呤的定量限和检出限分别为0.24~3.00和0.08~1.00 mg/kg。
2.4.2 加标回收率与精密度。选择适宜的已知嘌呤含量的SPI样品为研究对象,并根据样品嘌呤含量配制对应浓度的用于加标的混合标准溶液。按每种嘌呤本底值的0.5、1.0和1.5倍共3个浓度级别添加嘌呤标液,进行加标回收试验,由于样品中黄嘌呤为未检出(<3.0 mg/kg),则按其定量限的1、3和10倍的浓度级别添加嘌呤标液。每个浓度水平均重复制备6份,按“1.3”试验方法进行检测,得到相应的回收率和精密度结果。结果见表4,4种嘌呤的平均回收率为81.7%~95.2%,说明该方法的前处理条件可有效提取和保留样品中的嘌呤化合物,损失较少,分析干扰较小,准确度较高;4种嘌呤的相对标准偏差(RSD)为1.7%~8.6%,精密度良好,能满足一般检测要求。
2.5 实际样品分析
使用该试验优化的方法对10批不同来源的SPI样品进行4种嘌呤的测定分析,并将各组分加和计算总嘌呤含量。测定结果如表5所示,SPI样品中鸟嘌呤和腺嘌呤的含量较高,鸟嘌呤和腺嘌呤分别占总嘌呤的43.3%~58.8%和40.8%~55.5%,次黄嘌呤占总嘌呤的0.14%~1.21%,而黄嘌呤则均为未检出(<3.0 mg/kg),说明SPI中次黄嘌呤的含量非常低且几乎不含有黄嘌呤。10批SPI样品中总嘌呤的含量为1 236.6~3 636.9 mg/kg,平均值为2 209.9 mg/kg,与干黄豆的嘌呤含量(2 181.9 mg/kg)[12]接近。一般认为,食物中的嘌呤含量为 2 000~3 000 mg/kg时,属于较高组;嘌呤含量超过3 000 mg/kg时,属于非常高组[22]。这表明SPI属于嘌呤含量较高的食物,将其作为补充蛋白质的膳食食品原料或添加剂时,应充分考虑其用量和搭配,才能更有效地预防和控制高尿酸血症和痛风等疾病。
3 结论
该试验开发了同时测定SPI中鸟嘌呤、腺嘌呤、次黄嘌呤和黄嘌呤含量的超高效液相色谱-串联质谱方法。该方法简单快速、可操作性强,相比于主流的高效液相色谱法效率更高、选择性更强。对方法的标准曲线定量可靠性、定量限和检出限、准确度和精密度等参数进行了验证,结果表明该方法灵敏度较高、测定结果准确、回收率高、重复性好、满足一般检测要求。应用于实际样品的测定证实了其有效性和适用性,测试结果也显示SPI中嘌呤含量较高,属于高嘌呤食物。该方法为SPI在膳食食品中的科学应用提供参考依据,也为其他类型样品中嘌呤化合物及其类似物的测试提供了借鉴。
参考文献
[1] BAINY E M,TOSH S M,CORREDIG M,et al.Protein subunit composition effects on the thermal denaturation at different stages during the soy protein isolate processing and gelation profiles of soy protein isolates [J].Journal of the American oil chemists society,2008,85(6):581-590.
[2] 赵西周,梁少华.国内大豆分离蛋白生产现状与发展建议[J].中国油脂,2012,37(8):21-24.
[3] 时玉强,鲁绪强,马军,等.湿法粉碎豆粕对大豆分离蛋白生产的影响[J].中国油脂,2017,42(5):45-47.
[4] 常晨阳,常凯,朱秀焕,等.高效液相色谱-串联质谱法测定大豆分离蛋白中的三聚氰胺[J].现代农业科技,2018(21):252-253.
[5] 冯建岭,彭云婷,李迎秋.大豆分离蛋白的功能特性及应用[J].粮食与食品工业,2017,24(6):37-40.
[6] 戴卿印,周鑫,黄茜,等.大豆分离蛋白-壳聚糖可食用性抗菌膜的制备与性能评价[J].粮食与油脂,2022,35(6):89-95.
[7] 时玉强,马军.大豆分离蛋白生产质量控制探讨[J].安徽农学通报,2020,26(6):134-138,147.
[8] 陈佳俊,秦雪梅,杜冠华,等.基于嘌呤能系统及嘌呤代谢的抑郁症发病机制研究进展[J].药学学报,202 56(9):2464-2471.
[9] 金宗濂.嘌呤类物质生理活性和第三代保健(功能)食品研制与开发[J].食品科学,2000,21(12):200-205.
[10] 王奇,周衍衡,白占涛.痛风及其发病机制研究进展[J].延安大学学报(医学科学版),2018,16(4):82-85.
[11] 徐李华.食品中嘌呤含量与高尿酸血症关系的研究进展[J].中国城乡企业卫生,2020,35(1):26-28.
[12] 荣胜忠,张艳男,王栋,等.常见干豆类及豆制品中嘌呤含量研究[J].中国食物与营养,2014,20(6):61-63.
[13] 武惠敏,周雪巍,杨瑞,等.酵母蛋白中4种嘌呤含量的测定及对比分析[J].食品研究与开发,2023,44(10):177-185.
[14] 时玉强,牛祥臣,张彬,等.传统大豆制品及大豆蛋白中嘌呤含量的探讨[J].中国油脂,2020,45(7):61-66.
[15] 张庆,于晓章,张琳,等.微乳毛细管电动色谱-场放大富集法测定9种核苷类化合物[J].分析科学学报,2019,35(4):404-410.
[16] QU X,SUI J X,MI N,et al.Determination of four different purines and their content change in seafood by high-performance liquid chromatography[J].Journal of the science of food and agriculture,2017,97(2):520-525.
[17] 夏小乐,夏梅芳,杨海麟,等.LC-MS/MS法分析清爽型黄酒中的嘌呤含量[J].现代食品科技,2010,26(12):1399-1402.
[18] 彭松林,曾治国,张涛,等.肉类嘌呤含量及降嘌呤方法研究进展[J].食品与发酵工业,2022,48(18):314-321.
[19] 林洪,曲欣.食品中嘌呤含量分布的研究进展[J].食品安全质量检测学报,2012,3(5):373-378.
[20] 杨平,王瑶,宋焕禄,等.不同熬制条件下猪肉汤中滋味成分的变化[J].中国食品学报,2018,18(12):247-260.
[21] 孙玉凤,黄亚涛,刘佳萌,等.超高效液相色谱法检测58种大豆中的4种嘌呤化合物[J].中国食品学报,2023,23(6):304-313.
[22]蔡路昀,冷利萍,曹爱玲,等.食品中嘌呤含量分布研究进展[J].食品科学技术学报,2018,36(5):74-81.
