AlphaFold和蛋白质结构预测
2024-09-30郭贝一郭晓强
蛋白质是生命活动的物质基础,亦可看作生命的存在形式。蛋白质拥有广泛的生物学功能,包括结构组分(胶原蛋白)、催化功能(酶)、调节作用(激素)、物质运输(血红蛋白)、机械收缩(肌动蛋白)、机体免疫(抗体)等,进而参与几乎所有生命过程,如分子水平的DNA复制和转录、蛋白质翻译、物质与能量代谢等,以及细胞层面的精卵融合、细胞增殖和分化、细胞凋亡和坏死、细胞衰老和细胞通信等,其重要性不言而喻。
尽管蛋白质种类多样,但它们都是由20种基本氨基酸组成,区别在于不同的氨基酸排列顺序,这一特征称为一级结构,亦称初级结构,但这种线性结构需经过充分折叠形成空间三维结构,亦称高级结构。结构决定功能是许多学科的基本原则,生命科学也不例外,因此蛋白质结构研究在生命科学领域占据举足轻重的位置[1]。
蛋白质结构研究
1819年,构成蛋白质的第一种氨基酸亮氨酸分离成功,1936年最后一个氨基酸苏氨酸成功鉴定,前后跨越100多年,这说明了蛋白质研究的艰巨性。
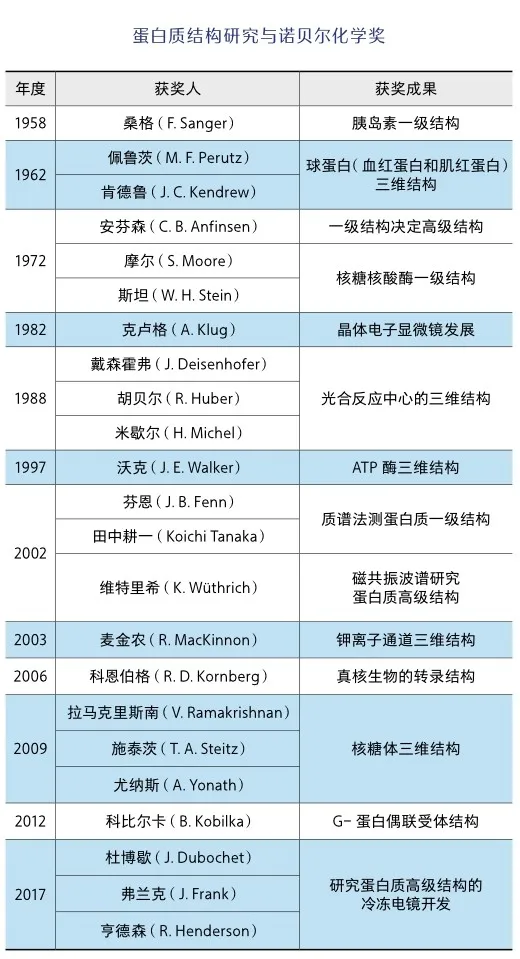
1953年,英国生物化学家桑格(F. Sanger)借助新出现的各种氨基酸和多肽分离方法和自己创造的氨基酸显色反应,确定了胰岛素两条多肽链的氨基酸序列,1955年又确定了二硫键位置,获得1958年诺贝尔化学奖。
1957年,英国生物化学家肯德鲁(J. C. Kendrew)借助X-射线晶体衍射技术首次确定肌红蛋白三维结构,两年后他的同事佩鲁茨(M. F. Perutz)确定血红蛋白三维结构,两人分享1962年诺贝尔化学奖。通过这两项诺贝尔奖也可看出蛋白质结构研究的重要性,从成果取得到获奖只有3~5年。与此对应,作为分子生物学里程碑成果的DNA双螺旋结构解析则等待了9年(如提前认可,富兰克林的悲剧可能就可避免)。
蛋白质结构研究一直是诺贝尔化学奖青睐的对象,至今已颁发十余次奖项,既包括重要蛋白(或复合物)的解析,也包括新技术或新方法的突破,因此该领域长期来看是生命科学前沿和焦点。
随着许多蛋白质结构被解析,科学界考虑将这些信息进行存档以便利科研人员使用。1971年,在美国冷泉港实验室举办的一次蛋白质晶体学研讨会上,由布鲁克海文国家实验室汉密尔顿(W. Hamilton)倡导建立蛋白质数据库(Protein Data Bank, PDB),最初只包含7种蛋白质结构信息,是生命科学领域第一个开放获取的数字资源。磁共振波谱和冷冻电镜等新技术的发明和完善,大大加快了蛋白质结构解析的步伐,PDB内信息也得到了快速增长,2014年突破了10万种。然而这一数量相比已知的上亿种蛋白质显得微不足道,因此研究人员开始尝试用理论方法直接由初级结构获取高级结构信息[2]。
蛋白质结构预测
1961年,美国生物化学家安芬森(C. B. Anfinsen)借助核糖核酸酶变性-复性实验得出结论:蛋白质初级结构决定高级结构(安芬森定律)。安芬森定律清晰地说明蛋白质空间结构并非随机形成,而是根植于氨基酸的线性顺序,该定律成为蛋白质结构预测的理论基础。安芬森在1972年诺贝尔奖获奖感言中提出一个愿景:将来有一天仅从氨基酸序列就能预测任何蛋白质的三维结构。从蛋白质初级结构预测高级结构的过程较为复杂,是结构生物学和理论生物学领域最具挑战性的课题,它吸引了众多科研人员前赴后继尝试解决这一难题。
蛋白质结构预测主要有两种策略,基于模板建模(template-based modeling, TBM)和无模板建模(template-free modeling, FM)[3]。TBM又称同源建模,它利用新蛋白质初级结构与PDB中已有三维结构蛋白质的氨基酸序列比对结果为基础构建模型,并进一步完善,准确性取决于新蛋白质和PDB中蛋白质的进化距离,如果PDB缺乏新蛋白类似结构域的已知结构,则无法生成准确模型。FM又称从头计算方法,利用能量函数计算氨基酸空间相互作用,最终从所有可能结构中选取最佳形式,依靠分子动力学模拟获得蛋白质折叠方式,该法对小分子量蛋白质结构预测还算准确,但随着蛋白质复杂性增加逐渐变得无能为力。
1994年,美国计算生物学家莫尔特(J. Moult)和菲德利斯(K. Fidelis)为推动蛋白质结构预测领域的快速发展,启动了蛋白质结构预测关键评估(Critical Assessment of Structure Prediction, CASP)活动,这是两年举办一次的竞赛或选拔赛,旨在评选蛋白质结构建模中的最新和最佳技术。组织者向参与者提供目标蛋白质的氨基酸序列,据此预测蛋白质结构,与此同时还采取实验方法获取蛋白质结构,最终将二者进行对比获得全局距离测试(global distance test, GDT)评分。为保证结果客观性,整个过程为双盲,即参与者和评审员互不知晓。GDT是百分制,用于评判结构预测的精准度,超过90分则非常理想,达到实验数据级别。在第一届竞赛中,当时的方法对在已知数据库有类似模板的蛋白质的理论预测尚算完美,但对无模板的则堪称灾难,GDT得分只有20分。即使到2016年第12届竞赛也只升高到40分,与预期还有极大差距,以至莫尔特悲观地认为,有生之年这个问题可能都难以解决,但不久人工智能(artificial intelligence, AI)技术的加入,使得问题得到了完美的答案。
人工智能发展
智能,又称智力,是人类所特有的复杂认知能力并在此基础上进行学习形成概念、理解和应用逻辑以及推理等。AI则是利用计算机或机器人模拟人类智能完成一系列复杂任务的过程,如推理等。
1950年,英国数学家图灵(A. M. Turing)在论文“计算机和智能”中首次讨论建造智能机器和测试智能的方式,赋予机器借鉴人脑信息加工、理性设计和科学决策的能力,奠定了AI的理论基础。1956年,在美国达特茅斯举办的学术会议上,麦卡锡(J. McCarthy)首次提出AI一词,并相信将来会制造出像人类思考和推理的机器。在随后的时间里,AI取得了一系列重要进展,特别是进入21世纪以来更是突飞猛进,智能水平日趋强大,这里重点介绍机器学习(machine learning, ML)。
机器学习是人工智能的分支,主要基于数据进行算法开发和研究,通过学习已有数据而推广到新数据,从而可在无明确指令情况下执行任务,进一步发展出更高级的深度学习(deep learning, DL),可在尽量减少人工干预的前提下实现对非结构化数据(如文本或图像)的学习,在视觉加工、语言处理和语音识别等领域得到了广泛应用。
深度学习的研究历史可追溯到1943年,美国神经生理学家麦卡洛克 (W. S. McCulloch)和年轻逻辑学家皮茨(W. H. Pitts)构建出第一个基于模拟人脑的神经网络(neural network)模型,它可以基于神经元的活动特性进行简单的逻辑运算。神经网络由多层结构组成,包含输入层、一个或多个隐藏层和输出层。只有三层的称为基本神经网络,三层以上的为深度神经网络,是深度学习的基础。
经过后续发展和完善,神经网络逐渐成为诸多AI设计的基础,人们开始尝试应用于蛋白质结构预测。比如2016年出现的循环几何网络(recurrent geometric network, RGN)和后续发展的神经能量模型和优化(neural energy modeling and optimization, NEMO)等,这些方法取得了初步成功。
天才少年
哈萨比斯(D. Hassabis)出生于英国伦敦,童年就展现出过人天赋。4岁时,哈萨比斯就能熟练掌握国际象棋技巧并战胜父亲和叔叔,13岁成为国际象棋大师,并在国际比赛中与成年人对弈。哈萨比斯还表现出与年龄不相称的逻辑运算天赋,8岁时应用电脑进行游戏编程,随着年龄增长,他决定将计算机应用于更高级的智力比拼,因此希望从事人工智能方面的工作。17岁时,他设计并编程了销量数百万、屡获殊荣的游戏《主题公园》。
1994年,哈萨比斯进入剑桥大学学习,3年后以年级第一名的成绩获得计算机学士学位。1998年,哈萨比斯创立电子游戏公司Elixir Studios,先后创作出《共和国:革命》《邪恶天才》等多款畅销游戏。
2005年,哈萨比斯在游戏领域证明自己的能力后,决定研究人脑机制以证明AI的巨大潜力,因此重返学术界,进入伦敦大学学院攻读认知神经科学博士学位,主要研究记忆和健忘的分子机制。哈萨比斯发现大脑海马区损伤会导致健忘症,并减弱联想功能和情景记忆,这项成果被《科学》周刊列为2007年十大科学突破之一。2009年,哈萨比斯获得博士学位后,前往美国哈佛大学和麻省理工学院开展博士后研究,进一步充实自己的知识。

2010年,哈萨比斯等人在伦敦创立人工智能公司DeepMind,将公司使命定义为解决“智能问题”,继而使用人工智能“解决其他一切问题”。哈萨比斯担任公司首席执行官(CEO),将自己在神经科学和机器学习方面的知识与计算机科学相结合,致力于构建新型人工智能机器。DeepMind的快速发展引起科技巨头谷歌的注意,并于2014年以超过65亿美元价格被谷歌收购,哈萨比斯仍担任DeepMind的CEO。
哈萨比斯认为围棋是机器学习的理想挑战目标,他们开发成功AlphaGo,在2015年以5∶0比分击败欧洲围棋冠军,第二年再以4∶1比分击败世界冠军李世石(Lee Sedol),证明了AlphaGo的强大能力。AlphaGo的成功也荣膺2016年《科学》周刊评选的年度十大科学突破之一。
人机对战的巨大成功促使哈萨比斯决定进一步挑战自身,这次不再关注游戏,而是生物学问题。哈萨比斯利用AI解决生物学问题的想法由来已久,但苦于找不到理想切入点而迟迟未能启动,蛋白质结构预测无疑是一个最佳选择。DeepMind为此成立了一个开展蛋白质结构预测的小团队,由于蛋白质空间结构的形成主要通过折叠(fold)方式完成,该团队开发的人工智能系统被命名为AlphaFold。
AlphaFold的首次突破
AlphaFold采用深度学习基础上的FM预测策略,共三个系统。首先是建模系统,利用神经网络和强大运算能力基于能量最低原理预测蛋白质内各氨基酸残基之间的空间距离,产生一系列结构片段(类似二级结构),初步组装出三维结构;其次是优化系统,对建模结构进行再计算的重复操作以提升精确度;最后是输出系统,产生最终三维结构。
2018年,AlphaFold参与了第13届CASP竞赛,牛刀初试就技压群雄,毫无争议地荣膺冠军宝座,复杂蛋白GDT平均达到60分,成功预测出给定的43种蛋白质中的25种,第二名仅预测出3种[6]。这项成就使哈萨比斯的工作第三次荣膺《科学》周刊评选的年度十大科学突破之一。
AlphaFold尽管较传统方法有较大突破,但仍存在诸多不足,如预测结果精确度尚待提高,距离90分以上(完美预测)仍有较大差距;预测结构平均分辨率为0.66纳米(大于0.3纳米时,仅显示蛋白质链的轮廓),与实验方法所得最佳分辨率0.1纳米(原子半径尺度,该分辨率下可清晰展示单个原子位置)也存在上升空间。
DeepMind公司决定改进AlphaFold系统,但经过6个月尝试后却远远未达预期,不得不开始重新调整思路,首先进行人事变动。
江珀(J. M. Jumper)出生于美国阿肯色州小石城,2007年在范德比尔特大学获得理学学位,随后在奖学金资助下进入剑桥大学攻读理论凝聚态物理学博士学位,但不久发现对此并无太大兴趣,因此选择退学并加入一家从事蛋白质结构计算机模拟的公司,工作中逐渐对理论生物学产生浓厚兴趣。2011年,江珀重新回到学校,进入芝加哥大学跟随著名理论化学家弗里德(K. F. Freed)和索斯尼克(T. R. Sosnick)攻读理论化学博士,将机器学习策略用于蛋白质动力学研究。2017年,江珀毕业获得博士学位,获悉DeepMind公司正在开展蛋白质结构预测方面研究,投递申请后很快得到答复,当年10月加入公司,成为蛋白质结构预测小组的重要成员。
在AlphaFold迟迟未能取得进一步突破之时,哈萨比斯意识到江珀在蛋白质物理和机器学习方面跨学科背景的重要性,他们进行了深入交流,最终达成共识,采用新思路改进原有设计。2018年7月,江珀被提拔为AlphaFold项目主管,全面负责总体工作。
AlphaFold2再度完善
江珀带领年轻团队对AlphaFold最初版本进行了重新梳理,在此基础上展开全面调整和改进,对每个细节给予挖掘以期达到尽善尽美。比如,他们引入空间立体结构和进化理念、整合已有蛋白质结构的详细信息如原子半径和键角等、完善机器有效学习策略以利于从有限数据中提取最大信息,特别是抛弃传统算法的束缚,更强调空间靠近而非线性相邻等。没有任何一种改进对最终结果有决定性影响,但正是这些奇妙新想法的完美结合,才最终实现真正意义的突破。
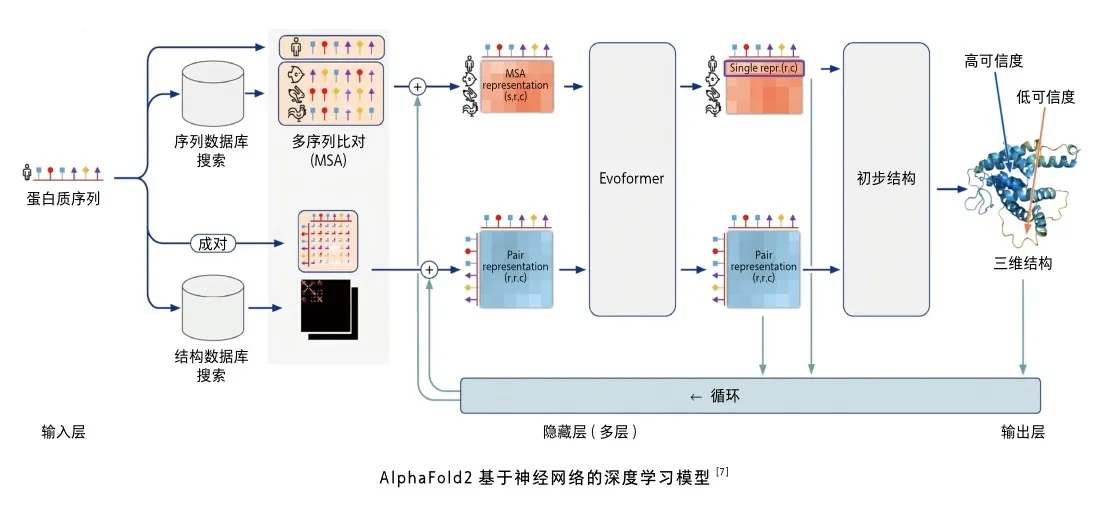
AlphaFold改进系统(亦称AlphaFold2)分为三个操作进程。第一进程,通过查询多个蛋白质的氨基酸序列数据库构建输入序列的多序列比对集(multisequence alignment, MSA)(MSA表征)。MSA的基本逻辑是同一功能蛋白质由于进化缘故,氨基酸序列会出现较大差异(如人肌红蛋白和鸽子肌红蛋白只有25%的氨基酸序列相同),但基本结构高度相似,据此作为结构预测的基础。同时,通过查询蛋白质三维结构数据库构建输入序列各氨基酸空间相互关系的矩阵(成对表征)。第二进程,上述两组信息(进化信息和空间信息)在一个编码器(evoformer)上进行处理,cB/Vi/goMYNWTFZcotgrOA==得出一个粗略的结构假设,然后返回最初状态进行测试和完善(该过程称为迭代),要求结构假设同时满足MSA表征和成对表征,且二者之间相互交流,共同纠正和改进最初假设结构的不足。第三进程是输出蛋白质三维结构。
深度学习过程中,研究者使用PDB中已有的17万种蛋白质结构信息进行训练。AlphaFold2反复将预测结果与真实结构进行对比,使二者之间逐渐接近,最终使系统吸收并完全掌握蛋白质折叠原理。AlphaFold2还能对预测结构给出可信程度,类似于考试估分,数值越高意味着与真正结构差距越小。AlphaFold2成功的另一因素是DeepMind强大的运算能力,这是绝大多数学术团体和小公司都难以达到的目标。
2020年,AlphaFold2在第14届CASP竞赛上进一步大杀四方,在给定预测蛋白质中GDT平均得分92.4,远超所有竞争对手;对高难度蛋白预测GDT平均得分87,比第二名高出25分。2021年7月15日,详细描述AlphaFold2内容的论文在《自然》周刊在线发表[7],至今引用近万次。DeepMind不久还公布算法供全球研究人员免费使用。这项成就被《科学》周刊评选为2021年度十大科学突破之首,蛋白结构预测也成为《自然-方法学》2021年度方法[8]。
AlphaFold2的发展和应用
AlphaFold2解决了蛋白质结构预测问题,目前虽无法做到尽善尽美,但大多数情况下对非结构解析专业的普通研究者而言已经足够。尤为重要的是,当研究人员获得感兴趣蛋白质序列时能够在几天甚至几小时内获得完美结构信息,而不再需要数月甚至数年时间和数百万美元的花费,对生命科学研究而言,就是难以置信的突破。
当然,AlphaFold2有不足之处,比如,对内在无序结构(它们在相分离等多个过程发挥重要作用)的预测效果较差。因为最初的设计目标是对单个蛋白质结构进行预测,但蛋白质发挥作用时往往是以复合物或小分子结合状态存在,为此,DeepMind公司和其他研究团队又开发出其他版本作为补充,如AlphaFold Multimer和RosettaTTAFold等。
AlphaFold2已展示出强大的蛋白质结构预测能力,2021年完成人类所有蛋白质的结构预测;2022年7月,获得2亿多种蛋白质的预测结构,并已进入数据库供免费使用[9,10]。DeepMind公司最近推出AlphaFold3版本,可高准确度和高精度预测蛋白质复合物、蛋白质-核酸、蛋白质与小分子配体等三维结构[11],进一步加大适用范围。
在基础研究领域,原来研究新蛋白功能需要依赖于X-射线衍射或冷冻电镜,高昂的费用和技术门槛使大多数实验室望而却步,目前该问题得到了有效解决。联合AlphaFold2和冷冻电镜成功解析核孔复合物结构这一成果对理解细胞核内外物质进出机制具有重要意义。
在应用研究领域,现代药物研发很大程度上依赖于蛋白质结构信息,但诸多感兴趣药物靶点蛋白尚未用实验方法解析出来,结构预测的突破无疑解决了这一瓶颈,必将极大推动新药研发进程。蛋白质人工设计如抗体和疫苗筛选是一个重要领域,传统方法是先设计出多种组合后,通过实验进行反复验证,过程烦琐耗时,但通过蛋白质结构预测则会大大缩短研究周期。
深远影响
蛋白质结构预测的突破可归因于天时、地利和人和的共同结果。天时上,高速发展的人工智能和层出不穷的新方法是AlphaFold的理论基础,PDB中快速增加的蛋白质结构是深度学习的材料基础;地利上,背靠谷歌公司强大的运算能力;人和上,公司聚集了一批富有朝气和探索精神的年轻人,以及哈萨比斯与江珀卓越的领导才能和创新能力,成功也就水到渠成。
近年来,AI在如火如荼地快速发展,取得一系列重大突破,如ChatGPT、Sora等,其智能程度逐渐提升,正在改变着人类的生活方式和科研的研究模式。AlphaFold无疑是优秀代表之一,可看作是AI发展的一个缩影。
AlphaFold解决了困扰生命科学多年的一个重大难题,并有望为其他生物学问题解决提供借鉴。大数据已成为当前科学发展的重要特征,如基因组测序结果和海量论文等,借助AI工具解决生命科学问题已成为一个重要方向。AlphaFold成功的另一层意义在于激励年轻人要敢于挑战自我。
AlphaFold2的成功也为哈萨比斯和江珀两位科学家带来众多科学荣誉。他们先后分享威利生物医学科学奖(2022年)、生命科学突破奖(2023年)、盖尔德纳国际奖(2023年)、拉斯克基础医学研究奖(2023年)等[12],也将是近几年诺贝尔自然科学类奖的热门人选。
(致谢:本文写作中获得周耀旗老师重要建议,在此表示诚挚的感谢。)
[1]Sklar J. QnAs with Demis Hassabis and John M. Jumper: Winners of the 2023 Albert Lasker Basic Medical Research Award. Proc Natl Acad Sci USA, 2023, 120(39): e2313816120.
[2]Rothman J E. Starting at Go: Protein structure prediction succumbs to machine learning. Proc Natl Acad Sci USA, 2023, 120(39): e2311128120.
[3]Kuhlman B, Bradley P. Advances in protein structure prediction and design. Nat Rev Mol Cell Biol, 2019, 20(11): 681-697.
[4]Zhou Y, Litfin T, Zhan J. 3=1+2: How the divide conquered de novo protein structure prediction and what is next? Natl Sci Rev, 2023, 10(12): nwad259.
[5]Jumper J, Hassabis D. The protein structure prediction revolution and its implications for medicine: 2023 Albert Lasker Basic Medical Research Award. JAMA, 2023, 330(15): 1425-1426.
[6]Senior A W, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature, 2020, 577(7792): 706-710.
[7]Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature, 2021, 596(7873): 583-589.
[8]Marx V. Method of the Year: Protein structure prediction. Nat Methods, 2022, 19(1): 5-10.
[9]Thornton J M, Laskowski R A, Borkakoti N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat Med, 2021, 27(10): 1666-1669.
[10]Borkakoti N, Thornton J M. AlphaFold2 protein structure prediction: Implications for drug discovery. Curr Opin Struct Bio, 2023, 78: 102526.
[11]Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024, 630(8016): 493-500.
[12]Sosnick T R. AlphaFold developers Demis Hassabis and John Jumper share the 2023 Albert Lasker Basic Medical Research Award. J Clin Invest, 2023, 133(19): e174915.
关键词:AlphaFold 蛋白质 结构预测 人工智能 ■
