硫代羧酸作为酰基化试剂在有机合成应用中的研究进展
2024-09-20王芸芸


摘 要: 硫代羧酸是重要的药物前体、化工原料及化学中间体,其独特的化学性使得该化合物在有机合成方面得到大力发展,如可作为酰基前体合成酰胺类衍生物。阐述了硫代羧酸作为酰基前体与含氮化合物在不同条件下进行酰胺化反应合成酰胺类衍生物的方法,对该方法存在的优缺点进行总结。
关 键 词:硫代羧酸;酰基前体;含氮化物;酰胺类衍生物
中图分类号:TQ216文献标识志码: A 文章编号: 1004-0935(20202024)0×8-1253-04
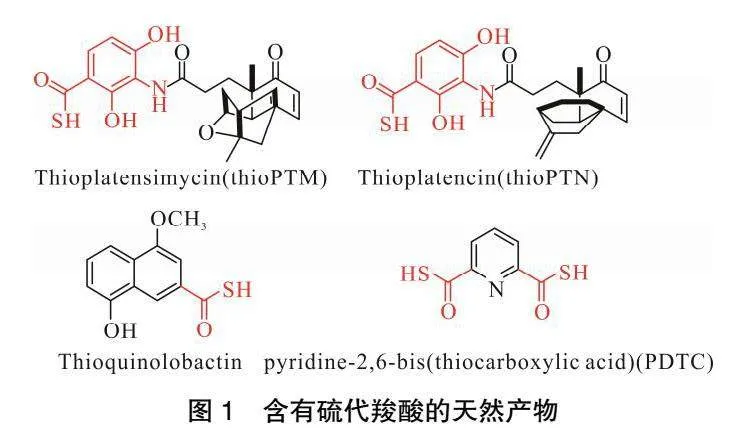
硫代羧酸[1],在药物合成、细胞代谢、生物修复等方面具有重要意义。在药物合成方面,如硫代平板霉素(thioPTM)和硫代平板素(thioPTN)可作为2种抗菌天然产物平板霉素(PTM)和平板素(PTN)的药物合成前体[2];在细胞代谢方面,如8-羟基-4-甲氧基-2-喹啉硫代羧酸[3]可作为生物降解催化剂;在生物修复方面,如吡啶-2,6-二硫代羧酸(PDTC)[4]可作为生物修复活性剂,如图1所示。此外,硫代羧酸及其衍生物也是重要的化工原料、添加剂[5]及化学中间体、还原剂[6-8],被广泛应用于化工、材料[9]等行业。
酰胺化是有机合成领域中重要的反应之一[10],酰胺化反应在药物合成、工业生产中有着广泛的应用[11-12]。传统的酰胺化反应是羧酸及其衍生物与胺的反应[13-14],该反应有的需要分子活化剂、有的需要高温等,反应条件较为苛刻。为实现更好的稳定性和原子经济性,探索和发展实用、绿色的酰胺化新方法具有重要意义。在过去的10年里,随着科学家的不断探索,硫代羧酸作为酰基来源在温和条件下形成酰胺键,越来越受到有机化学家的关注[15]。
1 无催化剂介导的硫代羧酸与含氮化合物的酰胺化反应
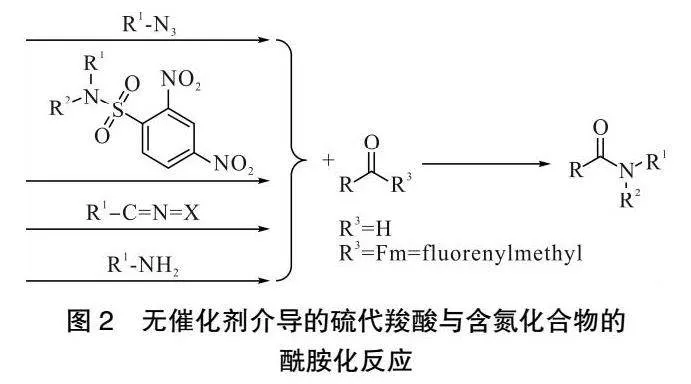
1988年,ROSEN课题组[16]报道了一种以硫代乙酸为酰基化试剂,与叠氮化合物在无催化剂条件下发生乙酰化反应,生成酰胺的方法。该反应过程中,叠氮化合物首先被硫代乙酸还原生成胺,然后胺与硫代羧酸偶联生成酰胺。该反应条件温和,化学选择性高,操作简单。
在此基础上,WILLIAMS课题组[17]于2003年报道了类似的反应。在实验过程中WILLIAMS等观察到噻三唑啉中间体,而不是叠氮化物的还原产物。他们认为噻三唑啉中间体通过[2+3]环加成或者重氮转移形成,后中间体通过逆[2+3]开环反应,最终分解为酰胺、氮和硫。该方法条件温和,没有副产物的生成。此外,利用该方法重氮化物可在不使用保护基团的情况下,在水溶液中制备传统方法难以获得的酰胺产物。
叠氮化合物难以制备,不容易保存,危险性高,故不是理想的胺源。于是1998年,TOMKINSON课题组[18]提出一种在无催化剂条件下,以碳酸铯作为碱,以DMF为溶剂,以 2,4-二硝基苯磺酰胺为胺源,与硫代羧酸反应生成酰胺化合物的方法。他们认为硫代羧酸与二硝基苯磺酰胺在碱的作用下生成中间体,后中间体脱去1分子二氧化硫得到酰胺产物和硫酚产物。该反应条件温和,酰胺产率高。其中,胺源硝基苯磺酰胺和二硝基苯磺酰胺容易制备,稳定性好。
2007年,CRICH课题组[19]报道了一种类似的酰胺合成策略。制备好的氨基硫代羧酸衍生物在碱的作用下,在DMF溶液中与2,4-二硝基苯磺酰胺反应生成氨基酸及多肽类化合物。此方法在一定程度上扩展了TOMKINSON课题组产物的应用范围。该反应生成氨基酸及多肽化合物的方法简便,反应条件温和,硫代羧酸前体(硫代羧酸衍生物)易于制备,且该前体与氨基酸保护基团相兼容,反应后得到高收率的酰化产物。
在此基础上,2009年CRICH课题组[20]发现一种类似的硫代羧酸前体,在室温条件下,与异氰酸酯或者异硫氰酸盐反应,以良好至优异的产率生成酰胺产物。他们认为硫代羧酸根离子,进攻异氰酸酯的SP碳,得到加成产物,后加成产物经过重排,脱去1分子COS或者CS2得到所需酰胺。
随着对硫代羧酸的不断探究,2019年HE课题组[21]开发了一种在无催化剂条件下硫代羧酸和胺构建酰胺键的方法。他们认为,二硫化物是合成酰胺的关键中间体。硫代苯甲酸可以在空气中自动氧化为二硫化物,硫代脂肪酸可以电氧化为二硫化物,生成的二硫化物与胺反应生成相应的酰胺。通过这种方法,可以在不使用任何催化剂或活化剂的情况下以优异的产率合成各种酰胺。生物活性化合物的成功合成也突出了这种策略在药物化学中的合成效用,如图2所示。
2 非金属催化剂介导的硫代羧酸与胺的酰胺化反应
随着异腈化学的不断发展,DANISHEFSKY课题组[22]开发了一种异腈参与催化的、硫代羧酸与胺类化合物进行双组分偶合(2CC)的新方法。他们认为异腈与硫代羧酸在室温下加成得到强酰基化中间体(thioFMAC),该中间体与胺进行偶联得到酰胺产物。这种新酰基化方法的优点是受空间位阻影响小,可以得到高收率的仲酰胺、叔酰胺。值得注意的是,这种化学方法也可很好地应用于酯的生成。
2011年,LIEBESKIND课题组[23]提出在温和的条件下,硫代羧酸与胺在N,O-双(三甲基硅基)乙酰胺(BSA)催化下,进行酰胺键和肽键构建的合成策略。他们认为硫代羧酸在BSA催化下生成S-三甲基硅基硫醇酯,而后S-三甲基硅基硫醇酯发生快速的硅化平衡,生成更稳定的O-硅基硫代酯,其与胺类化合物进行羧基化反应得到酰胺产物。通过这种硅基化化学反应,简单地描述了O-硅基硫代酯与胺的胺性和肽性偶联的关键反应原理,且该方法是早期报道中利用硫代羧酸在无金属催化下合成酰胺的经典方法之一。
XIAN课题组[24]报道了一种在室温或0 ℃条件下在THF溶液中,利用有机亚硝酸酯(RONO)或HCl/NaNO2对硫代羧酸进行活化,形成S-NO中间体,胺作为亲核试剂进攻S-NO中间体,形成酰胺产物的方法。他们认为该反应的硫化过程可以活化硫代羧酸,使得反应高效迅速进行。同样值得注意的是,与活化的硫代羧酸相较,NTA对羟基(如苯甲醇、苯酚和N-羟基琥珀酰亚胺)没有表现出任何反应性,这为选择性N-酰基化的进一步研究提供了希望。不足之处在于S-NO中间体是不稳定的,使得它的化学性质还没有得到很好研究。
2020年,BISWAS课题组[25]开发了一种3,6-二(吡啶-2-基)-1,2,4-5-四嗪(pytz)催化的硫代羧酸与胺进行高效、温和、无金属的直接酰胺化的合成方法。该方法适用于芳香族、脂肪族和杂芳香族硫代羧酸以及伯胺、仲胺、杂环胺甚至官能化的胺等。该反应具有官能团(如醇、酯、二硫代碳酸酯等)容忍性好、反应条件温和等优点,因此这种合成策略非常具有吸引力和实用性,如图3所示。
3 金属催化剂介导的硫代羧酸与含氮化合物的酰胺化反应
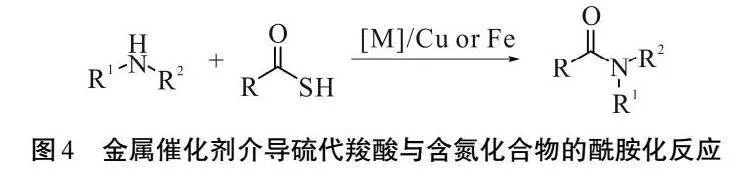
过渡金属催化是有机合成中比较常见的反应之一,利用过渡金属催化硫代羧酸酰化也得到有机化学家们的关注。2011年,GARNER课题组[26]发现一种在铜催化下,硫代羧酸与N杂环胺首先进行酰化,后在亲核试剂作用下生成无保护基团的肽链或氨基酸的合成策略。该方法反应条件温和,反应时间短,对无保护基团的肽链和氨基酸的合成具有重要的意义。
2013年,GOPI课题组[27]研究出一种硫代羧酸介导的,在温和条件下,氨基选择性N-酰基化反应的方法。硫代羧酸跟胺类化合物,在过渡金属铜的催化下,快速得到酰化产物。该反应条件温和,绿色,原子经济性高,反应迅速,短时间内得到的酰胺产物产率高。不足之处是在反应过程中,对强吸电子基团的耐受性差,即当存在强吸电子基团时,该反应活性降低。
2017年,MIRAKI课题组[28]发现磁铁矿纳米颗粒(Fe3O4@GAA - Cu(II))可以作为一种绿色、超顺磁性和可回收的纳米催化剂,应用于N-酰基化反应,得到所需酰胺产物。该反应条件温和、绿色,所需的催化剂是一种新型的纳米颗粒催化剂。在机理上该纳米催化剂的铜螯合部分与硫代乙酸相互作用,从而削弱了硫代羧酸的C—S键,使得硫代羧酸与胺类化合物在水中的酰化反应更加容易,如图4所示。
4 光催化剂介导的硫代羧酸与含氮化合物的酰胺化反应
相较于传统的化学合成方法,光氧化还原方法逐渐上升到合成有机化学的前沿,成为选择性小分子活化和化学键形成的一个不可或缺的工具[29]。在光照条件下,利用硫代羧酸作为酰基化试剂,与亲核试剂偶联得到目标产物,是有机化学家们不断追求的目标。
2016年,CHEN课题组[30]提出可见光介导的硫代乙酸钾与胺类化合物在光催化剂Ru的催化下进行有选择性酰化的合成策略。该反应方法不需要偶联试剂,反应条件温和,对官能团的容忍性高,且反应的化学选择性好。该反应所开发的合成策略具有通过二酰基二硫醚中间体来全面展示生物学上有趣的肽合成和氨基酸修饰的巨大潜力,具有很好的推广价值。
2017年,BISWAS课题组[31]发现在30 W的家用CFL条件下,在水溶液中,用新型CdS纳米颗粒催化剂催化硫代羧酸和胺反应,能够获得酰胺产物。该反应操作简单,条件温和,对胺上的官能团取代具有耐受性。这种多相光催化剂非常稳定,可以通过简单的离心过程被回收,而没有任何显著的催化性能损失。然而不足之处在于利用金属催化剂来转换光能,金属作为反应试剂它的引入会削弱了光氧化还原催化策略的优势。
2020年,SONG课题组[32]提出了一种经过可见光诱导,利用光催化剂催化硫代羧酸与苯胺发生酰化的合成策略。该反应表现出优异的化学选择性,而不影响其他官能团,如醇、酚、醚、酯、卤素或杂环等,还可进一步应用于温和条件下的氨基酸修饰,与水、空气兼容性好,以优异的收率获得酰胺产物,如图5所示。
5 结束语
酰胺键不仅是某些天然产物(如肽、蛋白质和几丁质)的重要骨架,也是有机化合物(如催化剂、药物、农药和材料)中最重要的官能团之一[33]。综上,科研工作者们已经掌握了多种利用硫代羧酸与含氮化合物进行酰胺化的方法来构建酰胺键。由此可知,硫代羧酸在有机化学中存在着巨大的应用潜力,寻求更加适宜的、绿色的、高效的方法来合成与硫代羧酸化学特性相关的化合物,仍需化学家们的不懈努力方向。
参考文献:
[1] ELAGAWANY M, HEGAZY L, ELGENDY B. Catalyst- and organic solvent-freezTDw/oWo1ue99Eb65vHPpetjwWtWZubwH6CKbn1Z1OU= synthesis of thioacids in water[J].Tetrahedron Letters, 2019, 60(30): 2018-2021.
[2] DONG L B, RUDOLF J D, KANG D D, et al. Biosynthesis of thiocarboxylic acid-containing natural products[J].Nature Communi- cations, 2018, 9: 2362.
[3] MATTHIJS S,TEHRANI K A, CORNELIS P,et al.Thioquinolobactin, a pseudomonas siderophore with antifungal and anti-Pythium activity[J].Environ. Microbiol.,2007,9:425-434.
[4] MATTHIJS S, BAYSSE C, KOEDAM N, et al. The Pseudomonas siderophore quinolobactin is synthesized from xanthurenic acid, an intermediate of the kynurenine pathway[J].Mol Microbiol, 2004, 52(2): 371-384.
[5] ALIEV I A, BELOVEZHETS L A, OPARINA L A. Fungicidal activity of S-esters of thiocarboxylic acids as antimicrobial additives to petroleum products[J].Petroleum Chemistry, 2019, 59(1): 99-105.
[6] KOBAYASHI F, FUJITA M, IDE T, et al. Dual-role catalysis by thiobenzoic acid in Cα—H arylation under photoirradiation[J].ACS Catal.2021,11:82-87.
[7] GAO C Z, FISHER Z B, EDGAR K J. Azide reduction by DTT or thioacetic acid provides access to amino and amido polysaccharides[J].Cellulose, 2019, 26(1): 445-462.
[8] LI Q, LUO Y, CHEN J, et al. Visible-light-promoted hydrogenation of azobenzenes to hydrazobenzenes with thioacetic acid as the reductant[J].J Org Chem, 2023, 88(4): 2443-2452.
[9] WANG J W, ZHAO H Y, XU L T, et al. Three-electron redox enabled dithiocarboxylate electrode for superior lithium storage performance[J].ACS Applied Materials & Interfaces, 2018, 10(41): 35469-35476.
[10] SANTOS A S, SILVA A M S, MARQUES M M B. Sustainable amidation reactions–recent advances[J].European Journal of Organic Chemistry, 2020, 2020(17): 2501-2516.
[11] UPADHYAY R, RANA R, MAURYA S K. Organocatalyzed C—N bond-forming reactions for the synthesis of amines and amides[J].ChemCatChem, 2021, 13(8): 1867-1897.
[12] DORR B M, FUERST D E. Enzymatic amidation for industrial applications[J].Current Opinion in Chemical Biology, 2018, 43: 127-133.
[13] PEDROOD K, BAHADORIKHALILI S, LOTFI V, et al. Catalytic and non-catalytic amidation of carboxylic acid substrates[J].Molecular Diversity, 2022, 26(2): 1311-1344.
[14] DAVIES J J, CHRISTOPHER BRADDOCK D, LICKISS P D. Silicon compounds as stoichiometric coupling reagents for direct amidation[J].Org Biomol Chem, 2021, 19(31): 6746-6760.
[15] N N, THIMMALAPURA V M, HOSAMANI B, et al. Thioacids–synthons for amide bond formation and ligation reactions: assembly of peptides and peptidomimetics[J].Organic & Biomolecular Chemistry, 2018, 16(19): 3524-3552.
[16] ROSEN T, LICO I M, CHU D T W. A convenient and highly chemoselective method for the reductive acetylation of azides[J].The Journal of Organic Chemistry, 1988, 53(7): 1580-1582.
[17] SHANGGUAN N, KATUKOJVALA S, GREENBERG R, et al. The reaction of thio acids with azides: a new mechanism and new synthetic applications[J].J Am Chem Soc, 2003, 125(26): 7754-7755.
[18] MESSERI T, STERNBACH D D, TOMKINSON N C O. A novel deprotection/functionalisation sequence using 2,4-dinitrobenzene- sulfonamide: Part 2[J].Tetrahedron Letters, 1998, 39(13): 1673-1676.
[19] CRICH D, SANA K, GUO S. Amino acid and peptide synthesis and functionalization by the reaction of thioacids with 2, 4-dinitrobenzene- sulfonamides[J].Org Lett, 2007, 9(22): 4423-4426.
[20] CRICH D, SASAKI K. Reaction of thioacids with isocyanates and isothiocyanates: A convenient amide ligation process[J].Org Lett, 2009, 11(15): 3514-3517.
[21] TANG L, MATUSKA J H, HUANG Y H, et al. Amide synthesis from thiocarboxylic acids and amines by spontaneous reaction and electrosynthesis[J].ChemSusChem, 2019, 12(12): 2570-2575.
[22] RAO Y, LI X, DANISHEFSKY S J. Thio FCMA intermediates as strong acyl donors: A general solution to the formation of complex amide bonds[J].J Am Chem Soc, 2009, 131(36): 12924-12926.
[23] WU W, ZHANG Z, LIEBESKIND L S.In situcarboxyl activation using a silatropic switch: A new approach to amide and peptide constructions[J].J Am Chem Soc, 2011, 133(36): 14256-14259.
[24] PAN J, DEVARIE-BAEZ N O, XIAN M. Facile amide formationviaS-nitrosothioacids[J].Org Lett, 2011, 13(5): 1092-1094.
[25] SAMANTA S, RAY S, BHADURI S N, et al. 3, 6-Di(pyridin-2-yl)-1,2,4, 5-tetrazine (pytz) catalysed metal-free amide bond formation from thioacids and amines at room temperature[J].Tetrahedron Letters, 2020, 61(35): 152272.
[26] DYER F B, PARK C M, JOSEPH R, et al. Aziridine-mediated ligation and site-specific modification of unprotected peptides[J].Journal of the American Chemical Society, 2011, 133(50): 20033-20035.
[27] MALI S M, BHAISARE R D, GOPI H N. Thioacids mediated selective and mild N-acylation of amines[J].J Org Chem, 2013, 78(11): 5550-5555.
[28] MIRAKI M K, YAZDANI E, GHANDI L, et al. Mild and eco-friendly chemoselective acylation of amines in aqueous medium using a green, superparamagnetic, recoverable nanocatalyst[J].Applied Organo- metallic Chemistry, 2017, 31(11): e3744.
[29] MCATEE R C, MCCLAIN E J, STEPHENSON C R J. Illuminating photoredox catalysis[J].Trends in Chemistry, 2019, 1(1): 111-125.
[30] LIU H X, ZHAO L Y, YUAN Y F, et al. Potassium thioacids mediated selective amide and peptide constructions enabled by visible light photoredox catalysis[J].ACS Catalysis, 2016, 6(3): 1732-1736.
[31] DAS S, RAY S, GHOSH A B, et al. Visible light driven amide synthesis in water at room temperature from thioacid and amine using CdS nanoparticles as heterogeneous Photocatalyst[J].Applied Organo- metallic Chemistry, 2018, 32(3): e4199.
[32] SONG W, DONG K, LI M. Visible light-induced amide bond formation[J].Org Lett, 2020, 22(2): 371-375.
[33] MONTALBETTI C A G N, FALQUE V. Amide bond formation and peptide coupling[J]. Tetrahedron,2005,61:10827-10852.
