基于福井函数方法的芳杂环化合物亲电取代反应位点预测及图形可视化教学应用
2024-05-29李琳杨宝华张爱华
李琳 杨宝华 张爱华

摘要:基于福井函数方法,利用Gaussian和Multiwfn软件预测了常见芳杂环化合物及取代芳杂环化合物的亲电取代反应位点,使之以可视化图像形式展现并应用于基础有机化学的教学实践。
关键词:Gaussian软件;Multiwfn软件;福井函数;芳杂环化合物;反应位点;图形可视化
中图分类号:O641;O626文献标志码:A文章编号:1001-2443(2024)02-0129-07
引言
在基础有机化学教学中,有机化合物的化学反应是核心知识内容。理解各类有机化合物的化学反应和结构的关系、讨论化学反应中反应底物不同位点反应的难易是教学的主线。目前,在教学中为了更好地理解化学反应,主要运用两种方法对化学反应的反应位点进行分析解释,其理论性都比较强,需要学生掌握扎实的有机基础理论。一种方法是基于化学反应热力学和动力学的相关理论,对于一些机理研究比较成熟的反应,通过判断反应途径中间体的稳定性及所需能垒的高低來对反应位点发生反应的难易进行分析解释。另一种方法是基于各种有机结构理论分析反应物自身的结构特点,以及反应影响因素如进攻试剂的性质、反应温度、溶剂、催化剂等进行分析解释[1-4]。前者在理论计算上主要是基于过渡态理论,通过考察不同位点上反应途径能量势垒的高低,明确反应机理,进而理解反应的本质。这种理论研究方法较为复杂繁琐,计算非常耗时。后者基于很多化学反应中反应底物自身的结构往往是最重要的因素,其他效应的影响往往相对次要。在理论上发展出一些完全基于反应物自身结构特点、忽略其他影响因素,直接判断反应物不同反应位点反应难易的方法。这类理论方法很多,但原理各不相同,包括前线轨道理论、福井函数、双描述符、静电势、原子电荷、电子定域化函数、电子密度拉普拉斯函数、相对亲核/亲电性等众多方法。该类方法在计算和分析上都相对方便快捷,但也各有优缺点,在分析具体有机体系时,要选择出最合适的方法来对反应位点进行预测。除了这两类理论计算方法,预测反应位点最为准确可靠的方法是从头算动力学模拟方法,该方法可以将各种影响反应过程的效应全面地考虑。但该方法计算过于耗时,很难用于实际问题的讨论[5-6]。
本文理论计算采用的福井函数就是基于反应物自身结构特点来预测不同反应位点活性高低的方法,是概念密度泛函理论框架中的一个重要概念,是被广泛运用的预测化学反应位点的实空间函数。该函数可以分为三类,分别用于预测亲核、亲电和自由基反应位点。预测亲电反应的福井函数可以表示为:
其中,ρ(N)是分子净电荷为零状态下的电子密度函数,ρ(N-1)是体系电离一个电子后,净电荷为+1状态下的电子密度函数。通常认为,福井函数值越大的位点,其相应的反应活性也越大,且在福井函数计算结果的可视化等值面图上,该原子被福井函数等值面涵盖的程度也越大[5-9]。
福井函数的计算结果可视化展现后,不仅直观,而且更利于初学者理解和掌握。同时基于反应物自身结构解释化学反应难易的分析方法,也充分体现了“结构决定性质”这一有机化学知识内涵,是基础有机化学教学中理解反应位点发生反应难易的重要分析方法。随着化学可视化软件的不断涌现,通过理论方法判断、预测和解释化学相关问题,并使之可视化已成为科学研究领域的重要手段和强有力的工具,将其有效地运用于教学实践,亦具有可行性,也必将成为提升教学质量的一大助力[10-12]。
芳杂环化合物是具有芳香性的一类重要的杂环化合物,在有机化学教学中占有重要地位。亲电取代反应是芳杂环化合物的重要反应类型,不同位点发生亲电取代反应具有明显的区域选择性。教学中通常利用电子效应理论和共振论对其反应位点进行定性解释说明,理论抽象,学生理解和记忆都不容易,教学效果不理想。利用福井函数方法,能够从图形可视化角度体现不同区域因电子密度发生变化而参与反应的能力,能让学生更好地理解和掌握芳杂环化合物的亲电取代反应规律。文献[5]将一些取代苯化合物作为测试体系,比较了各种最常用的预测亲电位点的方法,福井函数方法对于单个邻对位定位基和双取代基的取代苯体系的预测,都好于描述静电效应的方法如静电势、以及基于Hirshfeld、Mulliken、NPA等方法计算原子电荷等。福井函数方法在预测单个间位定位基取代苯体系时表现不是很理想。但本文用于具有邻对位和间位定位基的取代芳杂环化合物的预测,结果均可供借鉴。且该法计算量小,便捷易操作,尤其是用于教学实践,谨慎使用,能发挥很好的教学参考价值。
本文利用Gaussian 09[11-13]和Multiwfn软件[11-12,14]对常见芳杂环化合物和取代芳杂环化合物的电子结构进行理论计算和分析,并探讨了此类化合物的电子结构与亲电取代反应位点的关系。先在Gaussian 09程序包中利用密度泛函理论(density functional theory,DFT)在B3LYP/6-311+G(d,p)水平上进行中性体系(电荷数为0)的结构优化,然后使用相同的几何结构分别计算中性体系(电荷数为0)和电离一个电子(电荷数为+1)状态时的单点能任务,同时利用Gaussian命令out=wfn输出wfn格式的波函数文件。然后在Multiwfn波函数分析程序中先打开中性体系的wfn格式波函数文件,利用程序交互页面命令,先生成格点文件,并设定自定义运算,读入中性体系的波函数,之后再输入电荷数为+1状态的波函数文件,选择电子密度函数,对体系进行两个状态下的密度差的计算,之后为了得到合适的等值面图,可以进行格点质量的设置,并最终得到等值面图像[7]。等值面图像将抽象的理论计算结果以可视化图像的形式展现,直观形象,将其应用于有机化学的教学过程中,和现有的理解反应位点的有机教学方法相结合,搭建有机基本理论和应用的桥梁,利于学生更深刻地理解和应用抽象的有机化学理论。
文中所涉及的福井函数等值面图,均为研究体系发生亲电取代反应的图像,通过调节使等值面涵盖区域在不同原子上体现出差异,等值面值均设为0.005。f-函数值越大,表明f-越正,某个区域的原子被绿色区域涵盖越大,越容易发生亲电反应;蓝色区域与之相反,与f-负值相对应。
1 基于福井函数方法分析芳杂环化合物的亲电取代反应位点
1.1 含一个杂原子的五元单杂环化合物
图1是含一个杂原子的五元单杂环化合物吡咯、呋喃和噻吩的福井函数等值面图。吡咯、呋喃和噻吩是富电子体系,易于发生芳香亲电取代反应。由图1可见,吡咯、呋喃和噻吩在α位的福井函数电子密度较大,因而亲电取代反应更容易在α位发生。文献[1]认为,在发生亲电取代反应时,吡咯类的氮原子、呋喃环上的氧原子和噻吩环上的硫原子具有强的给电子共轭效应,会使亲电试剂进入杂原子的邻位和对位,对于五元杂环而言,应该是α和β位。但是在吡咯、呋喃和噻吩中,α位比β位活泼,因此反应实际上更易于在α位上发生亲电取代反应[1-2]。
文献[1]中利用前线轨道理论,从反应中间体稳定性角度,对α位比β位活泼这一说法与杂原子的定位效应导致的反应位点选择性不完全一致进行了解释说明。这对课程教学目标未达到这一难度和深度要求的非化学专业学生来说,理解起來比较困难。而利用福井函数的可视化图像直观展现不同反应位点发生亲电取代反应的难易差别,面对专业培养目标不同的学生,都有助于理解记忆。
1.2 含取代基的五元单杂环化合物
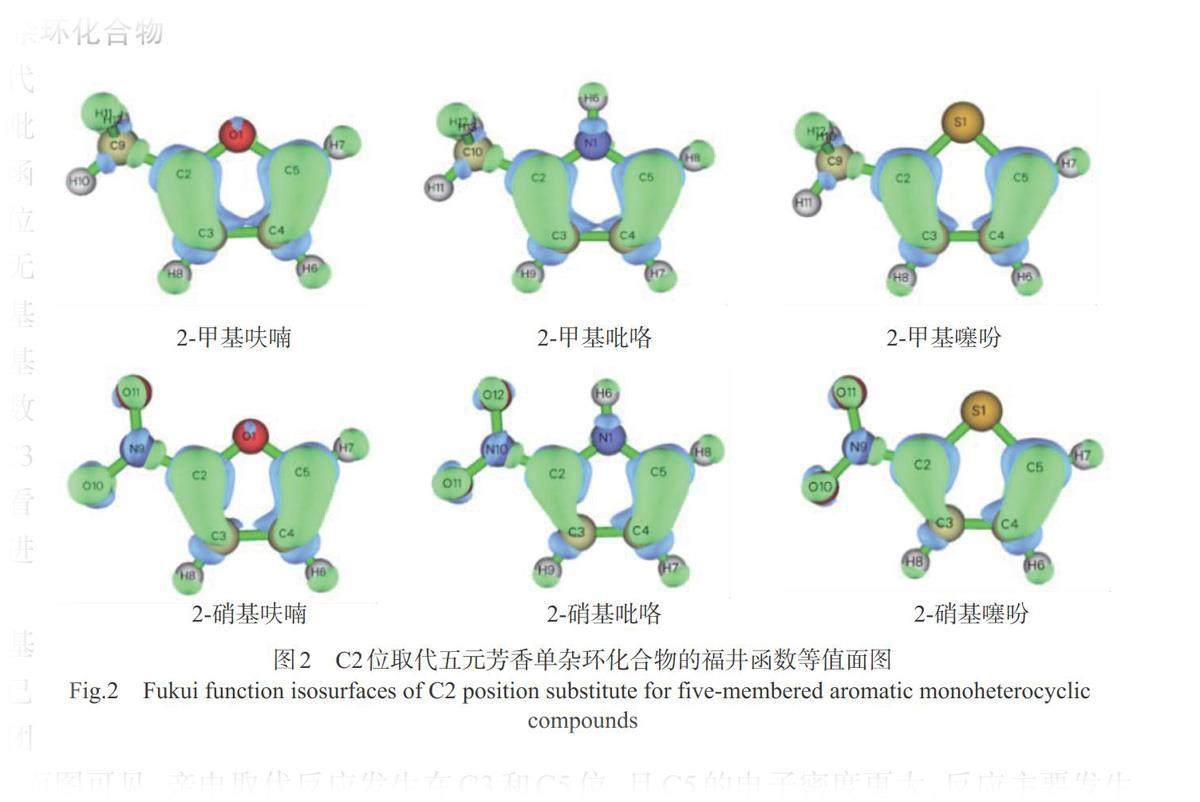
图2为C2位存在取代基团的五元芳香单杂环吡咯、呋喃和噻吩的福井函数等值面图。对于C2位已有取代基团的呋喃,无论取代基是邻对位定位基团的甲基还是间位定位基团的硝基,C5位福井函数表示的电子密度都比C3位和C4位密度大,可以看出新进入的取代基团易进入C5位。
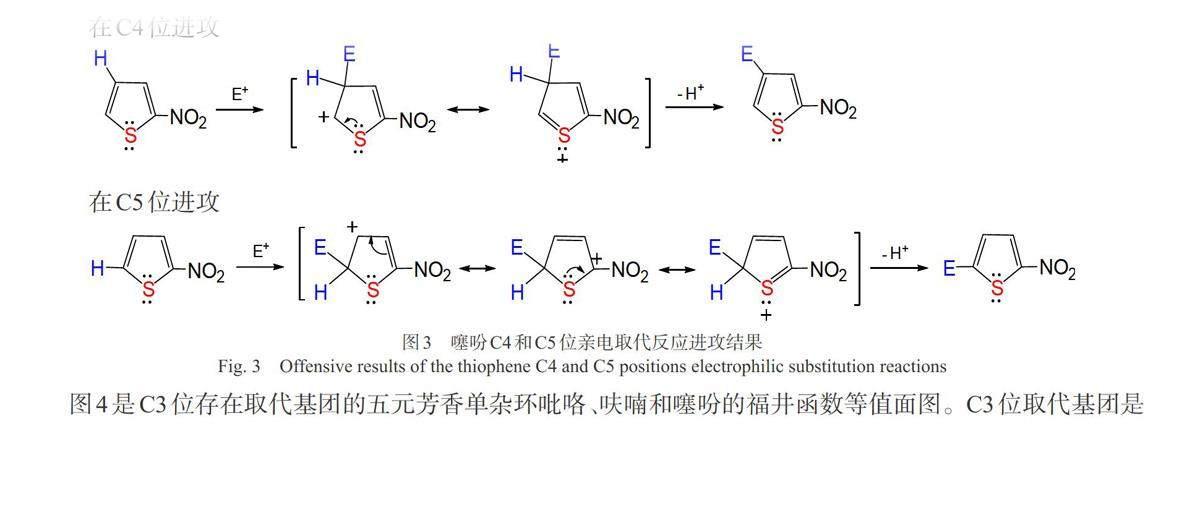
而C2位已有取代基团的吡咯和噻吩,如果已有基团是邻对位定位基团甲基,由福井函数的等值面图可见,亲电取代反应发生在C3和C5位,且C5的电子密度更大,反应主要发生在C5位上。当吡咯和噻吩的C2位已有基团是间位定位基团硝基时,反应发生在C4和C5位,且主要发生在C5位。福井函数等值面图很好地展现了N或S杂原子强的给电子效应和硝基的间位定位效应一致时的区域选择性。但文献[1]认为吡咯和噻吩的C2位取代基是间位定位基团时,反应发生在C4和C5位,且主要发生在C4位,而不是C5位。福井函数预测结果与文献结果稍有偏差。图3从共振论角度例举了噻吩C2位已有取代基团为间位定位基硝基时,C4和C5两种反应模式亲电进攻的所有可能的共振式。亲电试剂进攻C5位时比进攻C4时多产生一个共振式中间体,提示C5位比较有利于取代,但同时C4位对亲电进攻也是活泼的,且进攻C5位有一个共振式将正电荷置于带有吸电子硝基的C2位上,因此反应发生在C4和C5位上,这一点福井函数预测结果与之相同。文献[1]认为反应主要发生在C4位上,这极有可能是因为反应条件和亲电进攻试剂的影响使得C4位的产率提高所致。
图4是C3位存在取代基团的五元芳香单杂环吡咯、呋喃和噻吩的福井函数等值面图。C3位取代基团是邻对位定位基团甲基时,三个甲基取代化合物C2和C5位的福井函数电子密度均较大,且C2位更大,所以亲电取代反应更容易发生在和甲基相邻的C2位。C3位取代基团是间位定位基硝基时,三个硝基取代化合物C2和C5位的福井函数电子密度也较大,C5位更大,发生亲电取代反应时,后进入的取代基团主要进入硝基的间位C5位。利用福井函数预测并以可视化图像直观地展现出杂原子和定位基的定位效应导致的反应区域选择性。
1.3 六元单杂环吡啶衍生物
图5为取代吡啶的福井函数等值面图。吡啶是一个缺电子体系,如果在吡啶环上还存在一个吸电子的间位定位基团,那么发生亲电取代反应会更加困难,反而容易发生亲核反应。故此处只讨论吡啶环上的取代基是给电子的邻对位定位基团时化合物发生亲电取代反应的情况。
由图5可见,如果在C2位有给电子基团甲基或氨基时,亲电取代反应的主产物为C3或C5位的取代产物,C5位福井函数电子密度更大,取代产物可能会更多一些;如果在C4位有给电子基团甲基或氨基时,处于对称等价位置的C3和C5位的福井函数电子密度较大,亲电取代反应的主产物为C3或C5位的取代产物。预测结果与文献结论符合较好[1]。
图5表明,当吡啶C3位有给电子基团甲基和氨基时,亲电取代反应主要发生在福井函数电子密度较大的C6和C2位。但文献[1]认为,当C3位有强的给电子基团时,亲电取代反应主要发生在C2位,并未提及C6位。在这一点上福井函数预测的反应位点与甲基和氨基作为邻对位定位基影响亲电取代反应区域选择性的结果相一致,具有合理性,值得商榷。
1.4 含两个杂原子的五元单杂环化合物
图6为含有两个杂原子的五元单杂环吡唑、咪唑、噁唑、异噁唑、噻唑及异噻唑的福井函数等值面图。图示结果表明,两个杂原子的定位效应对亲电取代反应位点反应难易的影响非常明显。
由于亲电试剂或基团通常会在芳环相对富电子的位点进行反应,因此唑类芳杂环化合物的亲电取代反应位点取决于吡咯类氮原子或氧原子或硫原子[1]。由图6可以看出,1,2-唑类的异噻唑的亲电取代反应主要发生在C4位,吡唑和异噁唑的亲电取代反应位点利用福井函数展现出C4和C5位是首要反应位点;1,3-唑类的福井函数等值面图能明显地展现出亲电取代反应位点为C4、C5位。
这与文献[1]中利用共振论以中间体正离子极限式的稳定性对亲电取代反应发生区域选择性的解释基本一致,但利用福井函数计算并以可视化的等值面图的形式展现出来更直观、更形象生动。
1.5 常见的稠杂环化合物
图7为一些常见稠杂环化合物的福井函数等值面图。吲哚、异吲哚和苯并呋喃是苯环并五元杂环稠合的杂环化合物,图7清晰展现出3个化合物福井函数的最大电子密度均出现在五元杂环上,芳香亲电取代反应易于在杂环上发生。吲哚C3位的福井函数电子密度最大,表明吲哚的C3位最容易发生亲电取代反应。异吲哚和苯并呋喃福井函数展现出的亲电取代反应易于发生在对称的等价位置C1或C3位上。
文献[1]认为吲哚的亲电取代反应过程可理解为烯胺与亲电试剂的反应过程,如图8所示。当吲哚的亲电取代反应在C3位进行时,苯环的芳香性不变,亚胺正离子可以离域到苯环的π体系中,反应的能垒比较低;而在C2位反应时,苯环的芳香性被打破,此外亚胺正离子无法被芳香环稳定。因此,亲电试剂主要在C3位上进行反应。利用福井函数预测的结果与文献[1]中关于吲哚C3位易于发生亲电取代反应的理论解释一致,且更直观形象,易于理解。
喹啉和异喹啉的基本化学性质是吡啶和苯环的结合体,其芳香亲电取代反应主要发生在苯环上。由图7可以看出,喹啉的亲电取代反应易于发生在福井函数电子密度较大的C5和C8位,这一点和文献结论一致[1,4]。但异喹啉的福井函数等值面图虽然把首要亲电取代反应位点C5和C8分析出来了,但明显吡啶环的C1、C3和C4位造成了干扰,直观不容易区分。这一点可能与福井函数方法主要着眼于反应底物自身结构对反应位点的影响这一局限性有关,而实际实验结果受很多因素影响。这时也可以采用更合适的理论方法对反应体系进行预测和解释,文献[12]曾运用其他理论方法对喹啉和异喹啉体系进行过分析探讨,结果与实验结论符合得较好。
1.6 六元单杂环吡啶
六元单杂环吡啶因为氮原子比碳原子电负性大,氮原子通过吸电子诱导和吸电子的共轭效应使得环上碳原子的电子云密度更低,在图9所示的吡啶的亲电反应福井函数等值面图上,氮原子附近福井函数电子密度最大,可以解释吡啶的氮原子具有碱性且易于与强的亲电性介质成盐。另外,可以看出吡啶环上的电子云有向氮原子移动的趋势,体现出了氮原子的强的吸电子诱导和吸电子共轭效应。吡啶不是一个好的芳香亲电取代反应底物,但却是一个好的芳香亲核取代反应底物,在强烈的反应条件下,吡啶的亲电取代反应会在C3位发生[1]。但等值面图对吡啶C3位亲电取代位点展现得并不好,在图9中看起来不如C4位的等值面涵盖的程度大,一个原因可能是吡啶发生亲电取代反应需要强烈的反应条件,且发生亲电反应的难易受反应中间体稳定性的影响更大,受反应物自身电子结构的影响是次要因素;另一个原因可能是福井函数的亲电反应理论预测方法对于亲电反应倾向较弱的体系预测不准确。
因而,对于具有较弱亲电取代反应活性的反应底物,利用福井函数预测亲电取代反应位点时,如何增加预测结果的准确性和可靠性,还需要在理论和实践中进一步检验和探索。
2 结语
基于福井函数方法,利用Gaussian和Multiwfn软件对常见芳杂环和取代芳杂环体系的亲电取代反应位点进行了预测分析。福井函数方法对大部分由反应物自身电子结构起决定作用的芳杂环体系的亲电取代反应都有较好的预测结果,将之以可视化图像形式呈现出来,用于教学实践,更深刻地阐释了“结构决定性质”这一教学主线的逻辑关系。但运用这一理论方法预测反应位点时,有几个方面需要思考:一是由于每一种方法涉及的理论都较为复杂并存在局限性,对分子性质预测时并不总能成功[15]。福井函数方法也必然存在不足,但它在描述反应物结构与反应性关系时忽略了其他因素干扰,在定性描述物质结构与反应性关系方面具有较强优势,值得借鉴。二是由于实際化学反应体系的复杂性,部分体系发生化学反应时除与反应底物本身的性质密切相关以外,其他因素如进攻试剂的性质、空间位阻、反应温度、溶剂以及催化剂等的影响有时候也不可忽略,由此可能会导致理论预测与实验结果存在一定偏差,实际应用时需要注意。三是预测反应位点的理论方法很多,但不同方法基于的原理不同。在对反应位点进行预测时要注意选择合适的理论方法并注意评估其可靠性和准确度是否理想,以期减小理论方法和实验结果间的偏差。总之,在运用理论方法预测解释化学反应位点并进行教学实践时,要综合考虑,同时要结合相关文献和实验事实评估其可靠性和准确度。
在基础有机化学教学实践中,“结构决定性质”是有机化学的知识内涵,基于化合物自身的结构特点分析其具有的化学性质是教学的基本理念和思路,也充分体现了有机结构理论中结构和反应性之间的关系。探索利用可靠的理论方法对化学反应位点发生反应的难易进行预测分析,并使之直观可视化,在教学实践中会具有很强的说服力,助力教学质量的提升。
参考文献
[1] 邢其毅,裴伟伟,徐瑞秋,等. 基础有机化学[M]. 4版. 北京:北京大学出版社,2019: 5.
[2] 陆阳. 有机化学[M]. 9版. 北京:人民卫生出版社,2018: 11.
[3] VOLLHARDT K P C, SCHORE N E, 有机化学:结构与功能:下册[M]. 戴立信,席振峰,罗三中,等,译. 北京:化学工业出版社,2022: 1.
[4] 李兴海,张杨,秦培文. 基础杂环化学[M]. 北京:化学工业出版社,2019: 2.
[5] 付蓉,卢天,陈飞武. 亲电取代反应中活性位点预测方法的比较[J]. 物理化学学报,2014, 30(4): 628-639.
[6] 曹静思,陈飞武. 芳香化合物亲核、亲电反应活性的理论预测和实验反应速率的相关性研究[J].有机化学,2016, 36:2463-2471.
[7] SOBEREVA. 使用Multiwfn作电子密度差图[EB/OL]. (2019-05-22)[2022-12-08] . http://sobereva.com/113.
[8] WANG B, RONG C Y, PRATIM K C, et al. A comparative study to predict regioselectivity, electrophilicity and nucleophilicity with Fukui function and Hirshfeld charge[J]. Theoretical Chemistry Accounts. 2019, 138: 124-132.
[9] SOBEREVA. 通过轨道权重福井函数和轨道权重双描述符预测亲核和亲电反应位点[EB/OL].(2022-05-24)[2022-12-08].http://sobereva.com/533.
[10] 柏一慧,贾似藐,王艺璇,等.计算化学在正丁醚制备实验教学中的应用[J].化学教育(中英文). 2022, 43(2): 107-113.
[11] 李琳,杨宝华,张爱华.Gaussian和Multiwfn 软件在判断蒽和菲芳香性上的应用[J].化学教育(中英文),2020, 41(24): 92-97.
[12] 李琳,杨宝华,张爱华.基于Gaussian和Multiwfn软件的喹啉和异喹啉的可视化教学研究初探[J].首都师范大学学报(自然科学版),2021, 42(3): 44-51.
[13] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09[CP]. Revision C. 01, Gaussian Inc, Wallingford, CT, 2010.
[14] LU T, CHEN F W. Multiwfn: A multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry,2012, 33(5): 580-592.
[15] 李强根,李宣映,毛双. 单取代苯环定位效应定量分析和图形可视化解释[J].大学化学,2019, 34(1): 108-115.
Prediction and Graphical Visualization Teaching Applications of Electrophilic Substitution Reaction Sites of Aromatic Heterocyclic Compounds Based on Fukui Function Method
LI Lin,YANG Bao-hua,ZHANG Ai-hua
(Yanjing Medical College, Capital Medical University, Beijing 101300,China)
Abstract: The electrophilic substitution reaction sites of common aromatic heterocyclic compounds and substituted aromatic heterocyclic compounds were predicted theoretically based on the Fukui function method by using Gaussian and Multiwfn software, which were presented as graphical visualization images and applied to the teaching practice of basic organic chemistry.
Key words: Gaussian software;Multiwfn software;Fukui function;aromatic heterocyclic compounds;reaction sites;graphical visualization
(責任编辑:王海燕)
