蒙药材蜀葵花质量标准研究
2024-05-23郭宝凤
岳 海,塔 娜,2,高 寒,2,郭宝凤,2*
(1.内蒙古自治区药品检验研究院,内蒙古 呼和浩特 010020;2.内蒙古自治区中蒙药标准研究重点实验室,内蒙古 呼和浩特 010020)
蜀葵花为锦葵科植物蜀葵Althaea rosea(Linn.)Cavan.(Malvaceae)的干燥花[1],蜀葵入药历史悠久,始载于《尔雅》,收录在《中药大辞典》、《本草纲目》、《全国中草药汇编》等医学著作。蜀葵花具有清热,利尿,利水,固精,止血。可用于尿闭,肾热,膀胱热,水肿,滑精,月经过多[2]。蜀葵花的质量标准目前收载在内蒙古蒙药材标准中,检验项目仅有性状、显微鉴别、理化鉴别,质量标准较低,无法有效控制其质量[3]。本研究采用性状、显微和薄层色谱鉴别法对蒙药材蜀葵花进行真伪定性鉴别,采用2020年版《中国药典》通则中收载的水分、总灰分、酸不溶性灰分、浸出物的方法测得,采用高效液相色谱法(HPLC)对其含量进行测定,为蒙药材蜀葵花的质量标准的提升提供可靠依据。
1 仪器与试药
1.1 仪器
ME5 型百分之一电子天平(赛多利斯);BSA型万分之一电子天平(赛多利斯);KQ-500DE型数控超声波清洗器(昆山舒美);DM 3000型显微镜(LEICA); U3000型高效液相色谱仪(赛默飞)。
1.2 试药
山奈素、槲皮素对照品均购于为中国食品药品检定研究院,山柰素对照品(批号:110861-202214,纯度97.4%);槲皮素对照品(批号:100081-201610,纯度99.8%);甲醇为色谱纯,水为高纯水,其他试剂均为分析纯。
1.3 样品
本研究所用蜀葵花药材,经我院郭宝凤副主任药师鉴定为锦葵科植物蜀葵Althaea rosea(Linn.)Cavan.(Malvaceae)的干燥花。样品信息见表1所示。

表1 样品信息
2 方法与结果
2.1 蜀葵花的性状鉴定
不同采集地区蜀葵花药材性状特点基本一致,描述为本品多皱缩卷曲,呈不规则圆柱形或扇形,长2~4.5 cm,直径1~2 cm。有的有花萼和副萼,花萼杯状,5裂,裂片三角形,长1.5~2.5 cm;副萼6~7裂,长5~10 mm,两者均呈黄绿色至黄褐色,并被有较密的星状毛。花瓣皱缩卷曲,紫红色或暗紫色,单瓣或重瓣,展平后呈倒卵状三角形,爪有长髯毛。雄蕊多数,花丝联合成筒状;花柱上部分裂成丝状。气微,味微苦。
2.2 蜀葵花的显微鉴别(粉末特征)
本品粉末红紫色。花粉粒圆球形,直径110~170 μm,表面具刺状雕文。非腺毛为星状毛,多破碎,完整者由3~9单细胞组成,单个细胞长80~700 μm。草酸钙簇晶甚多,直径10~20 μm。螺纹导管较多。见图1。

图1 蜀葵花花粉末特征图
2.3 薄层色谱鉴别
取本品粉末约1 g,加石油醚(60~90℃)30 mL,水浴加热回流40分钟,弃去石油醚,药渣挥干,加乙醇40 mL,超声处理40分钟,放冷,滤过,滤液蒸干,残渣加乙醇 1mL使溶解,即得供试品溶液。对照药材溶液的制备:取自制蜀葵花对照药材1g,同供试品溶液的制备方法制得蜀葵花对照药材溶液。吸取前述溶液各10 μL,分别点于同一硅胶G薄层板上;以三氯甲烷-甲醇-水(13:7:2)10℃以下放置的下层溶液展开,取出,晾干,喷以5%香草醛硫酸溶液,在110℃加热至斑点显色清晰,供试品色谱中,在与蜀葵花对照药材色谱相应的位置上,显相同颜色的斑点。见图2。
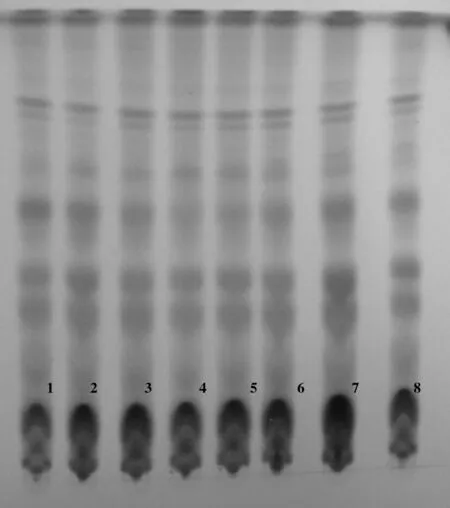
图2 蜀葵花薄层色谱鉴别
2.4 水分、灰分、浸出物含量测定
按照《中国药典》(2020年版)的方法,对7批蜀葵花样品的水分、总灰分、酸不溶性灰分进行检查,以70%乙醇为溶剂,按照浸出物测定法(通则2201)项下热浸法测定浸出物。结果见表2。
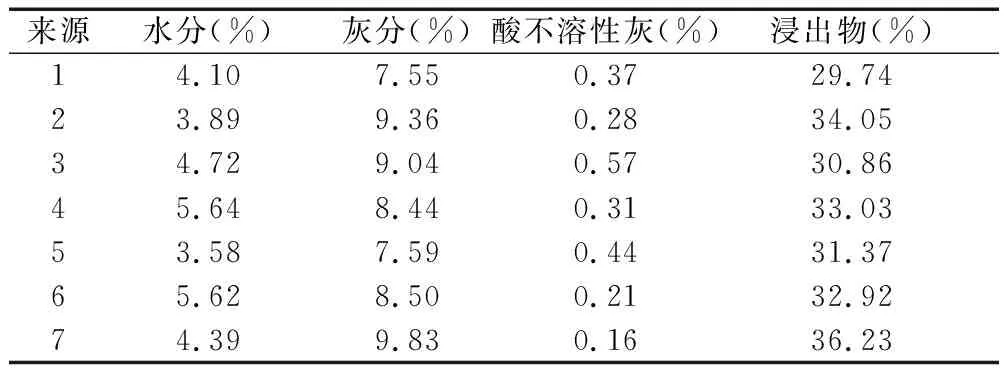
表2 水分、总灰分、酸不溶性、浸出物测定结果
2.5 含量测定
2.5.1 色谱条件[4]
C18色谱柱(250mm×4.6mm 5μm);以甲醇-0.4%磷酸(45:55)为流动相;流速为 1.0 mL/min;检测波长为360 nm;理论板数按山柰素色谱峰计算应不低于6000。
2.5.2 对照品溶液的制备
取山柰素对照品适量,精密称定,加80%甲醇制成每1mL含20 μg的溶液,即得。
2.5.3 供试品溶液的制备
精密称取取本品粉末约1 g,精密称定,置具塞锥形瓶中,加入80%甲醇50 mL,称定重量,水浴加热回流1 h,放冷,再称定重量,补足减失的重量,摇匀,滤过,取续滤液25 mL,置具塞锥形瓶中,加盐酸5 mL,置水浴中加热水解1 h,放冷,转移至50 mL量瓶中,加80%甲醇至刻度,摇匀,滤过,取续滤液,即得。
2.5.4 专属性试验
精密吸取对照品、供试品溶液各10 μL注入高效液相色谱仪。供试品色谱中,在与山柰素对照品色谱峰相同保留时间处出现色谱峰,且光谱图一致。表明该含量测定方法专属性好。以DAD检测器对供试品溶液中被测成分山柰素峰进行纯度检查,结果峰纯度为999.73。表明该方法测定蜀葵花样品中山柰素峰为单一成分。结果见图3。

图3 蜀葵花高效液相色谱图
2.5.5 线性关系考察
取山柰素对照品溶液(浓度为:21.62 μg/mL),分别以不同体积(2.0 μL、5.0 μL、10.0 μL、25.0 μL、30.0 μL)注入高效液相色谱仪,按“2.5.1”项下色谱条件进样测定,以峰面积对进样量做线性回归分析。结果山柰素在10.81 ng~135.13 ng范围内呈良好的线性关系。回归方程为:Y=0.0471X-0.0288,r=0.9999。
2.5.6 精密度试验(重复性)
取同一样品(2号)粉末(过三号筛)6份,各约1.0g,精密称定,分别按“2.5.1和2.5.3”项下条件进行测定,各进样10 μL,计算含量(mg/g),6份样品山柰素含量的 RSD 为1.58%,结果表明该实验方法、仪器性能稳定。
2.5.7 稳定性试验
取按“2.5.3”项下制得的同一供试品溶液,分别于0、2、4、6、8、12、24 h进行测定,记录峰面积,山柰素峰峰面积RSD为1.12%,表明该法制得的溶液在24 h内稳定性。
2.5.8 准确度试验
精密城称取重复性试验的样品(2号,2.729 mg/g)6份,每份约0.5 g,精密称定,置具塞锥形瓶中,分别精密加入山柰素对照品溶液10ml(浓度为0.1244 mg/mL),精密加80%甲醇40 mL,按上述方法测定,计算加样回收率,结果见表3。

表3 山柰素加样回收率试验结果
2.5.9 耐用性试验
更换三个厂家(岛津、安捷伦、迪马)的色谱柱,按上“2.5.1”项下的色谱条件,对同一份样品进行测定,分离度均大于1.5,测得的含量的RSD%为0.17%,表面该方法的柱耐受性好。
2.5.10 样品含量测定
分别称取7批蜀葵花样品,平行称2份,每份约1.0 g,按“2.5.3”、“2.5.1”项下方法测定7批蜀葵花样品中山柰素含量。结果显示,7批蜀葵花样品中山柰素含量为0.12%~0.49%,均值为0.26%,见表4。将蒙药材蜀葵花的限度暂定为按干燥品计算,含山柰素(C16H12O6)不得少于2.0 mg/g(0.20%)。

表4 蜀葵花中山柰素含量测定结果
3 讨论
3.1 薄层色谱鉴别
考察了甲醇、乙酸乙酯、丙酮和乙醇为溶剂超声提取法进行鉴别,结果色带干扰严重,无法准确检识,故采用石油醚脱色素后,再用乙醇为溶剂超声提取的方法、同时对比研究两种展开剂:乙酸乙酯-甲酸-乙酸-水(20:1:1:1)和三氯甲烷-甲醇-水(13:7:2)10 ℃以下放置过夜的下层溶液,对比研究两种显色剂:5%香草醛硫酸溶液和10%硫酸乙醇溶液,最终选取出分离度好,斑点信息全面的方法。
3.2 浸出物测定
照《中国药典》2020年版(通则2201)项下的水溶性浸出物测定法和醇溶性浸出物测定法,分别以水、30%乙醇、70%乙醇和乙醇为溶剂,进行试验研究,因蒙药材蜀葵花含有大量黏液细胞,水与30%乙醇溶液为浸出溶剂时,液体黏度大,过滤非常慢,无法操作,70%乙醇溶液比乙醇为溶剂时提取效率高,故以70%乙醇溶液为溶剂,热浸法测定浸出物含量。
3.3 含量测定中指标的选择
在含量测定试验中,同时以槲皮素、山柰素为检测指标进行研究[5-9],
结果供试品色谱中,虽然在槲皮素对照品色谱峰相同保留时间处有色谱峰出现,但光谱图不同,故未建立槲皮素的含量测定方法。在与山柰素对照品色谱峰保留时间相同位置处有出现色谱峰,且光谱图一致,建立山柰素的含量测定方法。
3.4 含量测定中供试品溶液制备方法
分别考察了不同提取方法(超声提取、回流提取、索氏提取)、不同提取时间(0.5h,1h,1.5h)及酸解效率考察(加盐酸3 mL、5 mL、7 mL)对蒙药材蜀葵花中山柰素的提取效率影响。结果确定最佳提取方法为80%甲醇加热回流提取1 h,酸解方法为加盐酸5 mL酸解1 h,提取效率最高。
