简便合成相可调的CsPbBr3-Cs4PbBr6 复合纳米晶及相转变过程的原位研究*
2024-05-13陈雪莲申岩冰袁芝聪李恺瑞潘喜强
陈雪莲 申岩冰 袁芝聪 李恺瑞 潘喜强
1) (西安石油大学材料科学与工程学院,西安 710065)
2) (西安石油大学化学与化工学院,西安 710065)
通过改变四正辛基溴化铵(TOABr)用量和Cs/Pb 摩尔比,在室温下采用一步单溶剂法成功制备出单斜相CsPbBr3 和六方相Cs4PbBr6 两种相结构可调的钙钛矿纳米晶.研究发现,当TOABr 浓度较低时(Cs/Pb/Br=1∶1∶4),体系中主要生成了单斜相的CsPbBr3 纳米立方块,该立方块主要经历了快速成核、尺寸分布聚焦生长和Ostwald 熟化生长3 个阶段,最终尺寸为(11.8 ± 1.6) nm.随着TOABr 用量的增加,Br-与Pb2+结合形成[PbBr3]-和少量的[PbBr4]2-络合物,两种络合物相互竞争.在成核期和生长早期体系中[PbBr3]-占主导,因而形成大量的CsPbBr3 纳米晶,随着反应的进行,体系中过量的Br-会与纳米晶中的Pb 相互作用,导致CsPbBr3纳米晶部分转变为具有六边形形状的Cs4PbBr6 纳米晶,同时[PbBr4]2-络合物的存在使得Cs4PbBr6 纳米晶继续长大,最终形成以CsPbBr3 为发光中心的CsPbBr3-Cs4PbBr6 复合纳米晶.只有当TOABr 用量为0.32 mmol时所得的CsPbBr3-Cs4PbBr6 复合纳米晶其光学性能和稳定性表现最佳.在此浓度下改变Cs/Pb 摩尔比只影响CsPbBr3 纳米晶和Cs4PbBr6 纳米晶在体系中的相对含量,当Cs4PbBr6 纳米晶含量较高时其荧光强度和稳定性相对较差.该工作对低温可控合成高效稳定的铯铅卤钙钛矿纳米晶提供一定思路.
1 引言
全无机铯铅卤钙钛矿纳米晶(CsPbX3(X=Cl,Br,I)PNCs)具有优异的光学性质[1-3],如长的载流子扩散长度、高的吸收系数、窄的光致发光、高的荧光量子效率和在可见光谱内的可调节带隙,使其在光伏、照明、探测和新一代显示领域展现出极大的应用潜力,也成为科研工作者关注的焦点材料.
自从2015 年Protesescu等[4]首次报道采用高温热注入(hot injection,HI)法成功合成出4—15 nm 尺寸可调的(CsPbX3(X=Cl,Br,I)PNCs纳米立方块,其发射线宽仅为12—42 nm、量子产率最高可达90%,荧光寿命为1—29 ns.随后,各种制备CsPbX3纳米晶的方法被相继报道,如室温配体辅助再沉淀法[5-7]、溶剂热法[8]、微波辅助法[9,10]、超声法[11,12]等.在采用这些方法合成CsPbX3纳米晶时,往往会添加有机配体-油酸和油胺作为纳米晶的表面稳定剂,由于CsPbX3纳米晶的较强离子特性以及配体与金属离子的弱相互作用,使得配体在纳米晶表面一直处于高度表面吸脱附状态,从而使纳米晶易团聚,导致钙钛矿纳米晶较差的稳定性[13-15].此外,在光、热、空气和极性溶剂存在下纳米晶的结构易遭到破坏发生分解,造成荧光猝灭.这些都严重阻碍了纳米晶在光电和光伏领域的工业化应用进程.因此,如何提高纳米晶的稳定性成为了目前亟待解决的重要难题.
围绕CsPbX3纳米晶稳定性差的问题,研究者们开展了大量研究工作,提出了稳定性提升策略,如配体的表面钝化[16-21]、无机物和高聚物的表面包覆[22-24].然而这些方法存在操作复杂、制备成本高和难以大规模生产的缺点.随着研究的不断深入,CsPbX3PNCs 衍生物—“零维”(0D)铯铅卤双钙钛矿纳米晶(Cs4PbX6(X=Cl,Br,I)PNCs)进入了人们的视野.与CsPbBr3PNCs 相结构不同的是,Cs4PbBr6材料中相邻[PbBr6]4-八面体被Cs+完全隔离,彼此互相独立,没有电子轨道的耦合作用.由于Cs4PbBr6与CsPbBr3的晶格匹配良好,CsPbBr3能原位转化成Cs4PbBr6,其余CsPbBr3可较好地嵌入在Cs4PbBr6基质中而形成CsPbBr3-Cs4PbBr6NC 复合结构,由于其独特的嵌入式结构,纳米晶的稳定性得到了有效提升[25-27].Xu等[28]报道了在采用阴-阳离子交换反应法制备过程中改变温度可获得一系列相占比不同的CsPbBr3-Cs4PbBr6复合纳米晶.研究发现,光致发光量子产率(PLQY)会随着立方相CsPbBr3纳米晶含量的增加而提高.Zhao等[29]以P2O5为成核剂,在碲锗酸盐玻璃里采用高温熔融淬火法制备了0-D Cs4PbBr6和3-D CsPbBr3相结构可调的复合纳米晶,不仅提高了纳米晶的储存稳定性,还有效提高了其光、热和水相稳定性.近期,Wang等[30]报道了将不同浓度的四正辛基溴化铵(TOABr)添加到溴化铯和溴化铅的前驱体溶液中,在室温下制备了相占比可调的CsPbBr3-Cs4PbBr6复合纳米晶.研究发现,复合纳米晶的荧光强度较CsPbBr3纳米晶有了明显提升.然而,目前的研究主要聚焦在CsPbBr3-Cs4PbBr6复合纳米晶的制备及性能方面,对其形成过程和相结构调控机制报道较少.
基于此,本文将分别以碳酸铯(CsCO3)、氧化铅(PbO)和四正辛基溴化铵(TOABr)作为独立的铯源、铅源和溴源,在只有油酸(OA)作为唯一配体的合成条件下,通过调节TOABr 的用量和Cs/Pb 摩尔比,成功在甲苯溶剂中制备出单斜相CsPbBr3和六方相Cs4PbBr6NCs 占比可调的CsPbBr3-Cs4PbBr6复合纳米晶.采用本课题组搭建的具有超高时间分辨能力的原位光致发光(PL)测试平台实时监测纳米晶的PL 光谱随时间变化曲线,剖析CsPbBr3NCs 的峰位、峰强、半峰宽的时间演变规律,对钙钛矿纳米晶的相结构调控机理进行初步探讨.
2 实验部分
2.1 药品与试剂
碳酸铯(Cs2CO3,99.9 %)、氧化铅(PbO,99%)、四正辛基溴化铵(TOABr,98%)均采购于Aladdin公司;油酸(oleic acid,OA,90%)采购于Sigma-Aldrich 公司;甲苯、乙酸乙酯采购于国药集团化学试剂有限公司.
2.2 铯铅溴钙钛矿纳米晶的合成
首先,将0.2 mmol Cs2CO3,0.4 mmol PbO 和2 mL OA 分别加入到样品瓶中,在磁力搅拌器上通过加热搅拌使得溶液变为透明.然后,称取一定量的TOABr 粉末,通过超声将其溶于2 mL 的甲苯溶剂中,配置出摩尔量分别为0.16 mmol,0.24 mmol,0.32 mmol,0.4 mmol 的TOABr 前驱体溶液.取0.2 mL 的含有Cs+和Pb2+的前驱体溶液加入到快速搅拌2 mL 的TOABr-甲苯溶液中,经过数秒反应后形成绿色溶液.为了去除多余的反应副产物及未反应的前驱体,向合成后的钙钛矿纳米晶中加入2 倍的乙酸乙酯反溶剂并在8000 r/min 下离心纯化,收集所得沉淀物,并将其分散在1 mL 甲苯中.
此外,将溴离子浓度固定为0.32 mmol 的条件下,进一步考察了Cs/Pb 的摩尔比(2∶1,1∶1,2∶3)对纳米晶的结构、形貌和光学性质的影响,纳米晶的制备过程同上,唯一不同的是PbO 的摩尔量(0.2 mol,0.4 mol 和0.6 mol).
2.3 样品的表征与性能测试
采用 X 射线衍射仪(XRD,德国布鲁克,D8 discover)对样品的晶体结进行表征;采用透射电子显微镜(TEM,日本电子,JEM-2100)进行样品形貌和尺寸表征;通过紫外-可见分光光度计(UV-vis,日本岛津,UV-2700)测量样品的吸收光谱;利用稳态荧光测试系统(PL,美国,Maya 2000 Pro)测量纳米晶的稳态荧光光谱.利用本实验室搭建的原位光致发光装置[21]监测了纳米晶的荧光光谱随反应时间的变化规律.每个光谱的时间间隔设置为100 ms,在连续监测80 s 后,停止搅拌,原位监测转为离线监测,继续连续监测72 h.
3 结果与讨论
3.1 Br-对纳米晶光学性质、相结构和形貌的影响
首先考察了Br-的用量对产物光学性质的影响,将Cs/Pb 的摩尔比固定为1∶1,通过改变Br-的摩尔浓度为0.16 mmol,0.24 mmol,0.32 mmol和0.4 mmol 来合成钙钛矿纳米晶.随后,结合PL 和UV-vis 探究了不同溴用量对反应80 s 后的纳米晶光学性质的影响,结果如图1 所示.图1(a)是对其进行荧光测试得到的PL 谱图,对数据进行归一化处理后可以清楚地看到,除Br-的摩尔量为0.4 mmol 时合成的样品其荧光峰出现在513 nm且峰形不对称外,其余溴用量下样品的荧光峰均表现出尖锐且对称的特点,这3 组样品随着溴用量的增加,荧光峰峰位依次为514 nm,516 nm 和516 nm.从未归一化的PL 谱图可以看出(见补充材料图S1(a) (online)),样品的荧光强度随着Br-摩尔浓度的增大出现先上升后明显下降的现象,当浓度为0.32 mmol 时荧光强度值最高.这4 个样品在自然光和紫外灯照射下的光学照片也显示(图S1(b)(online)),当浓度为0.16 mmol 时,样品在自然光下为浅黄色,紫外灯照射后发出淡淡的绿色荧光;随着Br-浓度的增大(0.24 mmol 和0.32 mmol),自然光下样品的黄颜色加深,而在紫外灯照射下发出亮绿色,0.32 mmol 时亮绿色更明显;当Br-浓度进一步提高到0.4 mmol 时,样品在自然光下的颜色接近乳白色,紫外灯照射下的荧光非常弱.通过对样品进行量子产率(PLQY)测试发现,纳米晶的PLQY 值可从Br-浓度为0.16 mmol 时的9%明显提高到Br-浓度为0.32 mmol 时的58%,然后显著降低到2%(0.4 mmol).说明Br-浓度在0.32 mmol 时样品呈现出较好的光学性能.
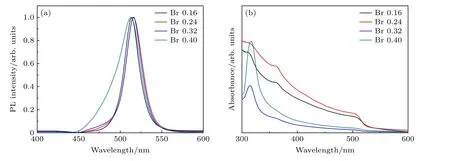
图1 不同TOABr 用量下所得铯铅溴纳米晶溶液的PL 光谱(a)和UV-vis 吸收光谱(b)Fig.1.(a) PL spectra (a) and UV-vis absorption spectra (b) of cesium lead bromide nanocrystal suspension at different dosages of TOABr.
图1(b)为纳米晶样品的UV-vis 吸收光谱图,4 组样品均在510 nm 左右出现CsPbBr3NCs 的特征吸收峰,但随着溴用量增加该吸收峰变得越来越不明显;与此同时,在314 nm 处出现典型的Cs4PbBr6NCs 特征吸收峰.这说明当Br-的用量≥0.32 mmol 时,同时存在CsPbBr3和Cs4PbBr6两种纳米晶的特征吸收信号,且随着溴用量的增加,源于Cs4PbBr6相的314 nm 的吸收信号会明显增强,表明体系中生成的Cs4PbBr6相的数量明显增加.已有大量文献报道[31,32],零维的Cs4PbBr6材料自身在绿光区无荧光发射,而PL 光谱观察到在516 nm 左右的发光可能源于零维六方相中的发射中心,也可能源于体系中存在的少量单斜相纳米晶.
随后,对4 个样品进行TEM 表征,以确定在不同溴用量下所得纳米晶的形貌,结果如图2 所示.图2(a)为溴用量0.16 mmol 时样品的TEM结果,图中只呈现出单一的CsPbBr3纳米立方块形貌,尺寸为(11.8 ± 1.6) nm;当溴用量增加到0.24 mmol 时(图2(b)),其样品形貌依旧为纳米立方块型,但尺寸减小到(9.6 ± 2.1) nm.而随着溴用量的继续增加(0.32 mmol 和0.4 mmol),图中以六边形形貌为主,表明有大量的Cs4PbBr6纳米晶生成[33],其六边形尺寸为(23.7 ± 2.3) nm,结果与UV-vis 相一致.仔细观察发现,六边形表面可清楚看见有较多小黑点分布(图2(c));而图2(d)中的六边形表面小黑点数量明显减小,六边形尺寸高达(66.1 ± 12.3) nm,尺寸均匀性也明显变差.为了明确这些小黑点的来源,对溴用量为0.32 mmol的样品进行HRTEM 表征,结果如图2(e)所示,这些黑点并不具备明显的衍射条纹.随后通过对它们的尺寸进行测量,如图2(f)显示其尺寸为(4.2 ±0.3) nm,这与Wang等[31]报道结果基本一致.因此,确定这些小黑点主要是源于体系中的CsPbBr3钙钛矿纳米晶(图中黄色圆圈区域),起到Cs4PbBr6NCs 发射中心的角色,这也解释了溴用量为0.32 mmol时样品的荧光发射能力较强的主要原因.
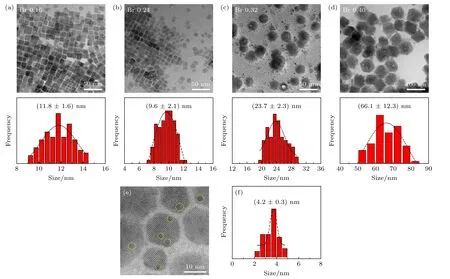
图2 (a)—(d)不同TOABr 用量下所得铯铅溴纳米晶的TEM 表征和晶粒尺寸统计结果,其中(a) Br 0.16 mmol;(b) Br 0.24 mmol;(c) Br 0.32 mmol;(d) Br 0.4 mmol;(e)为图(c)中选择的任意样品区域的HRTEM 图(黄色线圈为出现小黑点区域),(f)为小黑点晶粒的尺寸分布图Fig.2.TEM images and the corresponding histograms of cesium lead bromide nanocrystals synthesized at different dosages of TO ABr: (a) Br 0.16 mmol,(b) Br 0.24 mmol,(c) Br 0.32 mmol,and (d) Br 0.4 mmol.(e) HRTEM image of sample in panel (c) (the yellow circles represent the small black dots);(f) size distribution of the small black dots from panel (e).
通过XRD 对纳米晶的相结构进行了表征分析,结果如图3 所示,最顶部为Cs4PbBr6NCs 的六方相标准卡片(JCPDS No.73 -2487),最底部为CsPbBr3NCs 的单斜相标椎卡片(JCPDS No.18-0364),随着溴用量的增加,衍射峰的位置发生明显变化.在溴用量分别为0.16 和0.24 mmol 时,样品中仅能观察到单斜相CsPbBr3NCs 的特征衍射峰,分别为2θ=15.2°,21.2°,24.2°,30.4°,34.2°,37.6°,43.6°,依次归属于(001),(100),(110),(200),(120),(211)和(202)的衍射晶面.而在溴用量为0.4 mmol的样品中,分别在2θ 为12.8°,20.2°,22.5°,25.4°,27.5°,28.7°,30.4°,34.2°,39.1°出现明显的衍射信号,与六方相的特征衍射峰相吻合,说明此时主要以Cs4PbBr6NCs 为主,这些峰被归属于六方相的(110),(202),(113),(300),(024),(131),(214),(223)和(134)衍射晶面.但在溴用量为0.32 mmol 的样品中,同时出现单斜相和六方相的衍射信号,说明样品中同时存在两种相结构.为了更直观比较溴用量对所得纳米晶的相结构影响,对XRD 衍射峰采用面积法进行了计算[28],分别获得了CsPbBr3和Cs4PbBr6相的相对含量,结果如表1 所列.从表1可以看到,随着溴用量的增加,CsPbBr3相占比从96%逐渐减小到17%,而Cs4PbBr6相的占比逐渐增大到83%.更有趣的是,我们发现只有在溴用量分别为0.32 和0.4 mmol 的样品中出现了不属于单斜相CsPbBr3NCs 和六方相Cs4PbBr6NCs 的衍射峰,其峰位位于23.8°.经过与标准卡片进行对比,发现该峰属于PbBr2(JCPDS No.31-0679)的特征衍射峰.这说明,随着溴用量增大而出现的Cs4PbBr6,很可能是因为Pb2+与体系中Br-结合引起Pb2+被带出使体系成为贫铅环境所致.
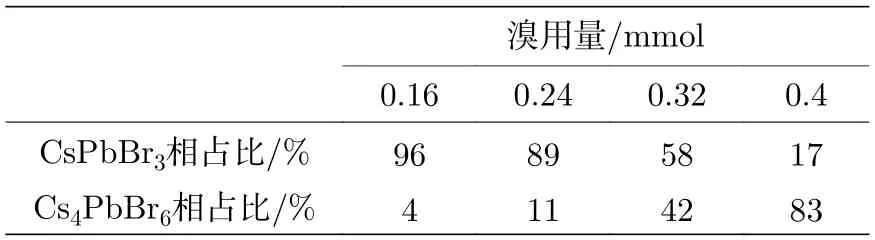
表1 不同TOABr 用量下所得纳米晶中单斜相CsPbBr3 和六方相Cs4PbBr6 的相占比Table 1. Proportion of CsPbBr3 and Cs4PbBr6 in nanocrystals synthesized at different dosages of TO ABr.

图3 不同TOABr 用量下所得铯铅溴钙钛矿纳米晶的XRD 图谱Fig.3.X-ray diffraction patterns of cesium lead bromide nanocrystals synthesized at different dosages of TOABr.
为了验证这一猜想,通过UV-vis 和PL 对体系中Pb2+与Br-的结合情况进行了考察.首先将0.4 mmol 的PbO 单独溶于油酸(OA),随后将Pb-OA 前驱体按制备纳米晶所需的相同量加入到TOABr-甲苯溶液中,充分搅拌混合后得到了Br-含量不同的4 组溶液,颜色均表现出淡黄色.PL结果显示,这些溶液都不具备荧光特性.随后,对这些溶液进行了UV-vis 测试,结果如图4 所示.可以看出,甲苯相的TOAB 溶液不存在吸收信号.而4 组溶液均在310 nm 出现明显的吸收峰,并且吸收峰强度出现明显不同,溴用量为0.16 mmol 的溶液吸收值最高,其余3 组溶液吸收值按从大到小依次为0.4 mmol,0.32 mmol 和0.24 mmol 的.同时,这3 组溶液在355 nm 左右还存在明显的凸起,其凸起程度会随着溴用量的增大而变强.结合文献[34,35]报道,确定了310 nm 处的吸收峰源于Pb2+与Br-结合产生的[PbBr3]-络合物,而355 nm处的峰则源于[PbBr4]2-络合物的吸收信号.该吸收信号变强反映了[PbBr4]2-络合物的含量会随着溴用量的增加而提高.实验结果表明,PbO 与TOABr 混合后非常容易结合,再结合XRD 中六方相样品中出现的PbBr2衍射峰现象,证明溴含量增加所导致的零维钙钛矿纳米晶生成,主要是PbO 与过量的TOABr 形成了PbBr2及铅溴络合物所致.
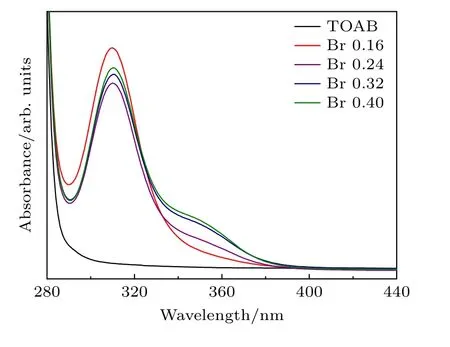
图4 不同浓度的TOABr 前驱体与Pb-OA 前驱体混合所得溶液的UV-vis图Fig.4.UV-vis absorption spectra of the solution obtained by mixing Pb-OA precursor and different concentration of TOABr precursor.
3.2 Cs/Pb 摩尔比对纳米晶光学性质、相结构和形貌的影响
通过改变TOABr 用量的实验发现,TOABr的浓度对钙钛矿纳米晶的相结构有显著调节作用,当TOABr 用量为0.32 mmol 时,生成的CsPbBr3-Cs4PbBr6纳米复合结构中单斜相和六方相的占比分别为58%和42%,此时样品的荧光强度较强,PLQY 值最高,展现出较好的荧光特性.将溴的用量固定在0.32 mmol,进一步考察了Cs/Pb 摩尔比对纳米晶相结构及光学性质的影响.
图5 给出了不同Cs/Pb 摩尔比下合成的钙钛矿纳米晶的PL 光谱和UV-vis 吸收光谱.从图5(a)的PL 谱图可以看出,随着Cs/Pb 摩尔比从2∶1逐渐增加到2∶3,所有样品均能观察到尖锐的荧光发射峰,峰位从517 nm 蓝移到512 nm 处,且荧光强度呈现出先增加后明显下降的现象,当Cs/Pb摩尔比为2∶2 时荧光强度最高.不同Cs/Pb 摩尔比对钙钛矿纳米晶的吸收光谱也有显著影响(图5(b)).当Cs/Pb 摩尔比<1 时,其吸收峰在314 nm 处出现,随后出现较长的拖尾,在510 nm 附近并未出现较为明显的CsPbBr3NCs 吸收峰;当Cs/Pb 摩尔比为1 时,除在314 nm 处存在Cs4PbBr6相吸收峰外,还在510 nm 左右出现较为明显的CsPbBr3结构的吸收信号;而当Cs/Pb 摩尔比>1 时,其314 nm 处的吸收峰较为尖锐且强度较高,但在510 nm 左右的吸收峰信号较弱.补充材料图S2(online)展示了不同Cs/Pb 摩尔比下合成的纳米晶液体样品在自然光下和紫外灯照射下颜色变化的光学图片.结果显示,Cs/Pb 摩尔比为2∶1 的样品接近乳白色,紫外灯照射下的荧光表现也相对较弱,而Cs/Pb 摩尔比为2∶2 的样品其颜色呈现出亮绿色,紫外灯照射下荧光颜色也很强,当Cs/Pb摩尔比为2∶3 时样品的颜色介于两者之间,这些变化规律与PL 结果相一致.
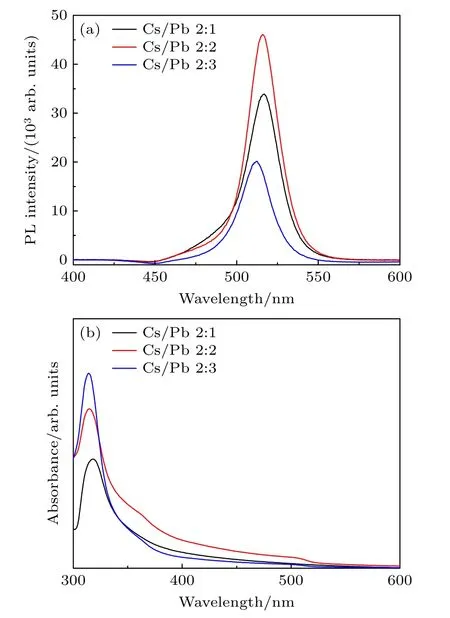
图5 不同Cs/Pb 摩尔比下所得铯铅溴纳米晶溶液的(a) PL光谱和(b) UV-vis 吸收光谱Fig.5.(a) PL spectra and (b) UV-vis absorption spectra of cesium lead bromide nanocrystal suspension at different molar ratio of Cs/Pb.
图6 为不同Cs/Pb 摩尔比下钙钛矿纳米晶的XRD 谱图.谱图中同样给出了单斜相钙钛矿(JCPDS No.18-0364)的标准卡片和六方相Cs4PbBr6型(JCPDS No.73-2478)的标准卡片.当Cs/Pb 摩尔比为2∶2 时,3.1 节已讨论过,此时存在单斜相和六方相共存,且两相之比为6∶4.当Cs/Pb 摩尔比为2∶1 时,分别在2θ=12.8°,20.2°,22.5°,25.4°,27.5°,28.7°,30.4°,34.2°和39.1°,这些峰被归属于六方相的(110),(202),(113),(300),(024),(131),(214),(223)和(134)晶面,确定其相结构主要为六方相.而当Cs/Pb 摩尔比为2∶3 时,两种相结构的特征衍射峰均存在,除上面提到的六方相的特征衍射峰外,还包括单斜相的2θ=21.2°,30.4°,37.6°和43.6°的衍射峰,依次归属于(100),(200),(211)和(202)晶面.与Cs/Pb 比为2∶2 时的结果相比,在相同测试条件下单斜相衍射峰的强度相对降低.为此,对不同Cs/Pb 摩尔比下所得纳米晶的相结构占比进行了研究,通过采用同样的面积法计算得到CsPbBr3和Cs4PbBr6相的相对含量,结果如表2 所列.这些现象说明,当Cs/Pb摩尔比<1 时,纳米晶更倾向于生成双钙钛矿相,而当Cs/Pb 摩尔比≥1 时,单钙钛矿相逐渐出现,相结构呈现出一种单斜相和六方相混合的结构,且当Cs/Pb 摩尔比为1∶1 时,单斜相的量最多、光学性能最好.
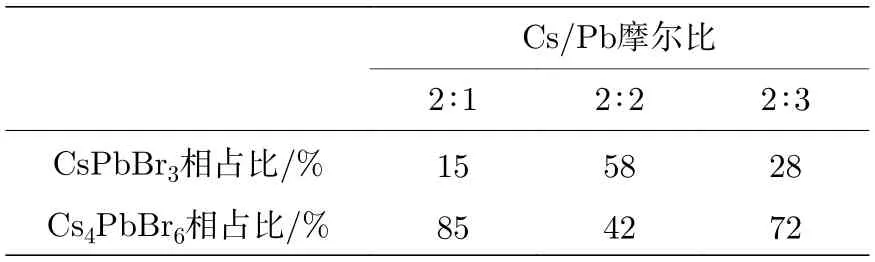
表2 不同Cs/Pb 摩尔比下所得纳米晶中CsPbBr3相和Cs4PbBr6 相的占比情况Table 2. Proportion of CsPbBr3 and Cs4PbBr6 in nanocrystals synthesized at different molar ratio of Cs/Pb.
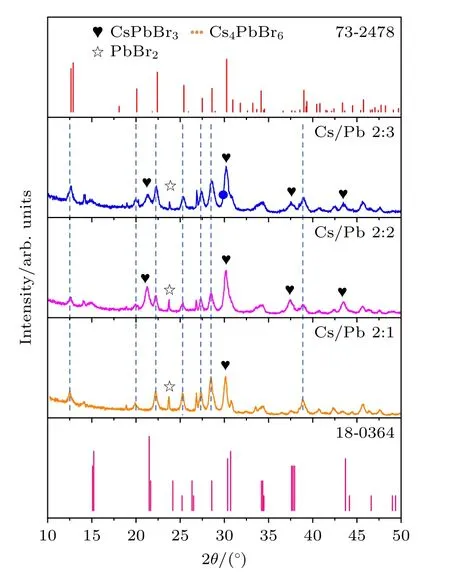
图6 不同Cs/Pb 摩尔比下所得铯铅溴纳米晶的XRD图Fig.6.X-ray diffraction patterns of cesium lead bromide nanocrystals synthesized at different molar ratio of Cs/Pb.
此外,还发现3 组样品中都存在源于PbBr2的23.8°衍射峰,为了验证Cs/Pb 摩尔比对Pb2+和Br-结合情况产生的影响,同样采用UV-vis 对Pb-OA 和TOABr-甲苯的混合溶液的吸收行为进行表征,结果如补充材料图S3 (online)所示.3 组溶液均出现[PbBr3]-络合物的310 nm 吸收峰,且随着铅量的增加,吸收值逐渐增大.而355 nm 处[PbBr4]2-络合物的吸收峰则呈现相反的规律,随着Pb 量的增大,吸收峰逐渐不明显.
图7 给出了不同Cs/Pb 摩尔比下合成的纳米晶样品的微观形貌图.其中图7(a)为Cs/Pb 比为2∶1 时所对应的TEM 图,样品的形貌主要为六边形的Cs4PbBr6NCs,其尺寸为(9.8±1.4) nm.当Cs/Pb 摩尔比为2∶2 时,如图7(b)所示,样品的形貌依旧为六边形Cs4PbBr6NCs,但尺寸增大到(23.7 ± 2.3) nm,还观察到Cs4PbBr6NCs 上分布着较多小黑点.而当Cs/Pb 摩尔比为2∶3 时,其形貌如图7(c)所示,虽仍保持六边形但却出现明显的团聚现象,尺寸为(22.5 ± 3.1) nm,小黑点数目明显较少.
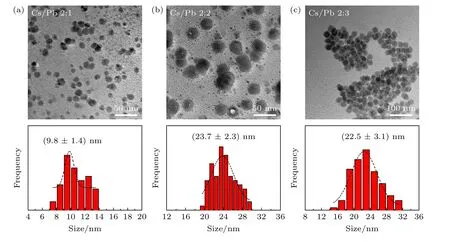
图7 不同Cs/Pb 摩尔比下所得铯铅溴纳米晶的TEM 表征结果和尺寸分布图(a) 2∶1;(b) 2∶2;(c) 2∶3Fig.7.TEM images and the corresponding histograms of cesium lead bromide nanocrystals synthesized at different molar ratio of Cs/Pb: (a) 2∶1;(b) 2∶2;(c) 2∶3.
3.3 不同Br-用量和Cs/Pb 摩尔比对纳米晶生长动力学的影响
3.1和3.2 节系统研究了TOABr 用量和Cs/Pb摩尔比对纳米晶的光学性质、相结构和形貌的影响,研究结果表明,调节Br-用量和Cs/Pb 摩尔比可以实现单斜相CsPbBr3NCs 和六方相Cs4PbBr6纳米晶间的相转变,当单斜相CsPbBr3NCs 占比为58%时样品的光学性能最优.为了深入理解纳米晶相转变机理,采用实验室搭建的原位PL 测试平台,实时跟踪了不同溴用量和Cs/Pb 摩尔比下所得纳米晶快速形成过程中PL 光谱的演变过程,将在线监测时间设置为80 s,每个样品点采集的时间间隔为100 ms,反应进行到80 s 后采用离线模式跟踪监测到72 h.
图8 为不同TOABr 用量下制备的纳米晶的原位PL 光谱,其中图8(a),(b)分别为TOABr 用量为0.16 和0.24 mmol 时合成的CsPbBr3纳米晶的原位PL 光谱(t< 80 s)和离线PL 光谱(插图).从图8(a),(b)可以看出,在0.1 s 时两样品都出现峰形对称的荧光发射峰,峰位分别在472 nm 和482 nm,此时荧光峰的强度较弱;随着反应的进行(0.1—80 s 之间),两个实验条件下荧光峰峰位逐渐红移到513 nm 和517 nm,同时荧光强度显著上升,而PL 峰峰形由不对称变得较为对称;随着反应时间的进一步延长(t> 80 s),峰位缓慢增大而峰强逐渐降低.当溴用量为0.32 mmol 时,如图8(c)所示,在t< 80 s 内,起始荧光峰位及峰位的红移规律与溴用量为0.24 mmol 时的相同;不同的是,当t> 80 s 时,PL 峰的峰位几乎保持不变且峰强下降较为缓慢.当TOABr 用量提高到0.4 mmol 时(图8(d)),纳米晶的起始荧光峰峰位值较大(490 nm)且峰形不对称,随后出现较快的荧光强度上升和峰位变化,在80 s 时峰位为513 nm,峰形仍旧不对称,直到420 s 后才呈现出对称性,而荧光强度则在较短的时间内迅速降低直至消失.
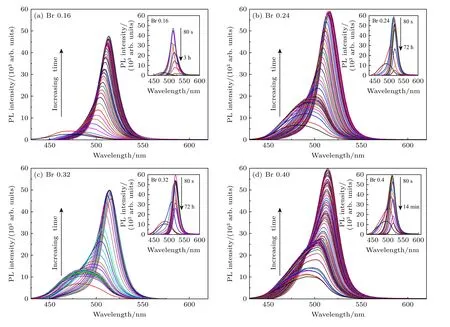
图8 不同溴用量下所得铯铅溴纳米晶在80 s 内的原位PL 光谱图,插图为纳米晶在80 s 后的离线PL 光谱图Fig.8.In-situ PL spectra of cesium lead bromide nanocrystals synthesized at different dosages of bromide ions within 80 s.The inset shows the ex-situ PL spectra of nanocrystals at reaction time after 80 s.
为了深入揭示不同实验条件对CsPbBr3和CsPbBr3-Cs4PbBr6纳米晶的形成动力学过程影响,对图8 的PL 谱图进行定量分析,分别获得了不同TOABr 用量下纳米晶样品的峰位、半高峰宽(FWHM)和峰强随反应时间的变化关系图,如图9(a)—(c)所示.根据荧光峰峰位、FWHM 和峰强随反应时间的变化规律划分为3 个阶段: 第1 阶段为刚开始出现的平稳期阶段,第2 阶段为峰位快速增大的阶段,第3 阶段为峰位几乎保持不变的阶段.在第1 阶段主要发生了前驱体的形核过程,通常晶核尺寸较小,数量少,会导致荧光强度较弱.结果显示溴用量不同会导致平稳期时间存在明显差异.其中在TOABr 用量为0.16 mmol 的条件下,铯、铅、溴三者的比值最接近1∶1∶3,此时纳米晶为单斜相CsPbBr3结构,在晶核快速生成的过程中,峰位与FWHM 几乎保持不变.平稳期在4 个样品中保持的最短,在稳定1 s 后,纳米晶的形成进入第2 阶段,荧光峰从472 nm 快速红移到511 nm,直到13 s 后才出现缓慢增长的趋势;同时FWHM 从42 nm 减小到22 nm,峰强也显著上升.这些现象表明晶核经历了快速长大的过程,且在该过程中立方块的尺寸分布迅速下降,说明CsPbBr3纳米晶在形成过程中经历了尺寸聚焦生长[36].随着时间的继续延长(t> 13 s),峰位会从511 nm 缓慢增至517 nm,反应进入到第3 阶段.FWHM 从22 nm 逐渐上升到28 nm,说明CsPb Br3纳米晶生长后期发生了Ostwald 熟化现象,使得纳米晶继续生长,尺寸分布明显下降,导致纳米晶的荧光强度出现明显衰减.

图9 不同溴用量下所得铯铅溴纳米晶的PL 峰峰位(a)、半峰宽(b)及峰强(c)随反应时间的变化规律图Fig.9.Changes in PL peak position (a),FWHM (b),peak intensity (c) of cesium lead bromide nanocrystals synthesized at different dosages of Br- as a function of reaction time.
当TOABr 用量为0.24 mmol 时(Cs/Pb/Br的摩尔比为1∶1∶6),XRD,TEM 和UV-vis 表征分析得知此条件下纳米晶样品主要为单斜相CsPbBr3结构,在第1 阶段(t≤10 s),与0.16 mmol 条件下不同的是,晶核形成过程中峰位值在缓慢增加,说明形核和生长过程在同时进行,以形核过程为主;随后峰位开始快速上升,荧光峰从487 nm 逐渐红移到514 nm,在此过程中FWHM 从50 nm 快速减小到23 nm,同时峰强出现明显上升现象.当t> 25 s 时,荧光峰峰位、FWHM 和峰强几乎保持不变.这些现象表明,随着TOABr 用量的增大,在第1 阶段CsPbBr3晶核的形成所需时间相对较长,同时伴有晶核长大现象;在第2 阶段(10 <t≤25),晶核会快速长大,纳米晶尺寸分布也更为均匀,纳米晶主要经历了尺寸分布聚焦生长模式;在第3 阶段(t> 25 s),没有发生Ostwald 熟化现象,说明TOABr 用量的增大提高了纳米晶的稳定性,抑制其继续生长,因而在反应结束后纳米晶的荧光强度值相对较高.
当TOABr用量≥0.32 mmol 时(Cs/Pb/Br 的摩尔比为1∶1∶8),经实验证实最终产物中会有六方相Cs4PbBr6NCs 的生成,且含量会随着TOABr用量的增加而显著增加.六方相Cs4PbBr6NCs 是如何形成的,对单斜相CsPbBr3纳米晶的形成过程有何影响? 随后分别对溴用量为0.32 mmol 和0.4 mmol 条件下合成的样品其峰位、FWHM 及峰强的时间演变规律图进行分析.结果发现,当溴用量为0.32 mmol 时,晶核出现的起始峰位位于482 nm,与溴用量为0.24 mmol 时的值相同,该峰位在13 s 内一直处于一个平稳期,其FWHM 值和峰强在平稳期略微上升,说明在此阶段有大量晶核形成且形核时间较长.由图4 可知,在反应初期,体系中同时出现了可形成单斜相的[PbBr3]-络合物和形成六方相的[PbBr4]2-络合物,两种络合物会相互竞争,导致CsPbBr3晶核形成时间较长.随后(13 s <t< 50 s),CsPbBr3纳米晶的生长过程和生长路径与0.24 mmol 时较为相似,只经历了尺寸分布聚焦生长,但生长过程持续到50 s.在CsPbBr3纳米晶生长过程中,由于TOABr 过量,其表面会出现TOABr 与Pb2+结合而生成PbBr2,因而会出现以CsPbBr3纳米晶为晶核而生长成复合型CsPbBr3-Cs4PbBr6纳米晶.
当溴用量为0.4 mmol 时,最终纳米晶产物中主要以六方相Cs4PbBr6结构为主(相占比为83%).此时,CsPbBr3晶核的荧光峰峰位变化过程与前3个实验条件下的略有不同,其起始峰位高达492 nm,说明TOABr 用量的增大导致CsPbBr3纳米晶的晶核尺寸显著增大,但形成晶核所需时间很短,仅约为2 s,略长于溴用量为0.16 mmol 条件下的成核时间.这可能是因为,在反应初期大量的TOABr与Pb2+结合形成PbBr2,使体系处于贫铅状态,体系中主要存在形成六方相的[PbBr4]2-络合物,而用以形成单斜相的[PbBr3]-络合物量较少.在第2 阶段(2 s <t≤ 30 s),CsPbBr3纳米晶晶核在逐渐长大,峰位会从492 nm 红移到510 nm,峰强也呈现逐渐上升趋势,上升趋势较慢,同时FWHM值则从50 nm 减小到35 nm,也呈现出缓慢下降的趋势;随着反应时间的继续进行(t> 30 s),峰位会继续增加到517 nm,而FWHM 从35 nm 继续下降到20 nm 以下,未出现平衡阶段,荧光强度则在达到峰值后突然消失.结合XRD 和TEM 表征,确定了体系中最终产物主要为六方相Cs4PbBr6纳米晶,其尺寸高达(66.1 ± 12.3) nm.结合图9(b)的FWHM 变化可知,过高的TOABr 浓度导致纳米晶的成核时间较短(~2 s),消耗掉的Cs4[PbBr6]单体量相对较少,更多的单体用于Cs4PbBr6纳米晶生长,较长的生长过程使得六方相纳米晶的最终尺寸较大.在最终产物中,CsPbBr3纳米晶的含量较低,当反应结束后荧光全部消失.
不同Cs/Pb 摩尔比下所得纳米晶的PL 光谱的时间演变关系图如补充材料图S4 所示(online).所有条件下,CsPbBr3纳米晶的PL 光谱的时间演变规律较为相似.在反应时间为0.1 s 时,当Cs/Pb摩尔比分别为2∶1,2∶2 和2∶3 时,CsPbBr3纳米晶的荧光峰起始峰位分别出现在474 nm,481 nm和476 nm;随着反应的进行峰位分别红移至517 nm,516 nm 和519 nm,同时强度逐渐上升,FWHM 明显变窄.对比发现,只有Cs/Pb 摩尔比为2∶2 时,CsPbBr3纳米晶的最终荧光强度最高,为Cs/Pb摩尔比为2∶1 时的1.6 倍,2∶3 时的2.5 倍.
同样,对图S4 的在线PL 谱图进行定量分析,获得了3 组不同Cs/Pb 摩尔比下纳米晶样品的峰位、FWHM 和峰强随反应时间变化的关系图,如图10(a)—(c)所示.CsPbBr3纳米晶的形成过程依旧可以划分为3 个阶段: 平稳期阶段(晶核形成期),峰位快速增加阶段(晶核生长期),峰位恒定阶段(停止生长期).当Cs/Pb 的摩尔比为2∶1 时,合成处于贫铅环境,更有利于六方相Cs4PbBr6结构的生成,晶核形成持续了约4 s,随后峰位开始增加,最后趋于平缓,整个变化规律与Cs/Pb 的摩尔比为2∶2 时的较为相似.不同的是,荧光峰的起始峰位位于474 nm 明显小于Cs/Pb 的摩尔比为2∶2时的481 nm,且FWHM 值为40 nm 明显低于Cs/Pb 的摩尔比为2∶2 时的51 nm,说明此条件下形成了尺寸较为均一的小晶核.但随着反应的进行,在Cs/Pb 摩尔比为2∶1 的样品里FWHM 又出现了短暂的上升后保持平稳.这种反常现象可能是在成核早期体系中生成了[PbBr3]-络合物和[PbBr4]2-络合物,但因体系中铅量较少,此时前体中还有大量的溴离子未被消耗,这些溴离子将会与[PbBr3]-络合物发生作用,产生[PbBr3]-+Br-=[PbBr4]2-的转换过程,当溴离子被消耗的差不多时,这种转换将会停止.同时,体系中的[PbBr4]2-络合物已经占据主导地位,将在Cs4PbBr6纳米晶表面继续沉积生长,同时FWHM 值也将持续下降,使纳米晶尺寸趋向于均一化.当与Cs/Pb 的摩尔比为2∶2样品的荧光强度进行对比时,发现虽然两者的最终荧光峰峰位保持一致,但Cs/Pb 的摩尔比为2∶1的样品的荧光强度明显偏弱,且在反应150 s 后出现下降现象,说明体系中的CsPbBr3含量明显低于后者,且随着反应时间的进行,CsPbBr3纳米晶不稳定,会逐渐转变为Cs4PbBr6结构.

图10 不同Cs/Pb 摩尔比下所得铯铅溴纳米晶的PL 峰峰位(a)、半峰宽(b)及峰强(c)随反应时间的变化规律图Fig.10.Changes in PL peak position (a),FWHM (b),peak intensity (c) of cesium lead bromide nanocrystals synthesized at different molar ratio of Cs/Pb as a function of reaction time.
当Cs/Pb 的摩尔比为2∶3 时,体系处于富铅条件,CsPbBr3纳米晶晶核的形成时间最短,仅为1 s,随后出现持续上升,直至达到最终稳定状态;而FWHM 在第2 阶段呈现的下降趋势较为缓慢,说明尺寸分布聚焦过程进行缓慢;在较长时间监测下(t≥ 80 s),FWHM 值反而增大,说明纳米晶在此时出现了明显的Ostwald 熟化现象,最终产物的荧光强度在3 个样品中最弱.结合TEM,XRD和UV-vis 表征结果,此时纳米晶主要为类六边形的Cs4PbBr6NCs,其尺寸较大,且出现了明显团聚现象,因而导致荧光强度最弱.
3.4 CsPbBr3 -Cs4PbBr6 NCs 纳米晶形成机理的探讨
当Cs,Pb 和Br 三种前驱体同时存在时,根据化学键理论,Cs—Br 键的键能为389.1 kJ/mol,Pb—Br 键的键能为248.5 kJ/mol,显然Pb—Br键更容易生成[35,37].将TOABr-甲苯溶液与Pb-OA 前驱体混合后,通过对Pb2+和Br-结合分析也发现了Pb—Br 键更易形成.随着Br-浓度的增大,溶液中除了生成[PbBr3]-络合物外,还会生成[PbBr4]2-络合物.根据文献[35,38]报道,[PbBr3]-络合物是CsPbBr3结构的基本骨架,最终产物CsPbBr3NCs 会由[PbBr6]4-八面体组成,它们通过在3 个正交方向上共享角来生成无限三维(3D)的[PbBr3]-框架;而[PbBr4]2-络合物则充当构成Cs4PbBr6NCs 的基本骨架.因此,结合实验结果,我们提出了不同合成条件下所得CsPbBr3NCs 和复合CsPbBr3-Cs4PbBr6NCs 相应的生长机理(如图11 所示).
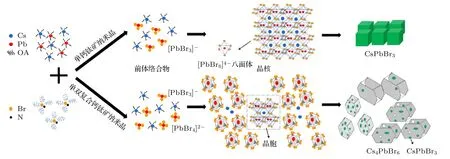
图11 CsPbBr3 纳米晶和CsPbBr3-Cs4PbBr6 复合纳米晶的生长机理图Fig.11.Schematic presentation of growth mechanisms of CsPbBr3 NCs and CsPbBr3-Cs4PbBr6 composite NCs.
1)当TOABr 用量较低时(≤0.24 mmol),体系中主要出现了大量的[PbBr3]-络合物,这些络合物会作为CsPbBr3的框架而快速形核,随后经历尺寸分布聚焦生长得到9—12 nm 的CsPbBr3NCs;随着TOABr 用量的逐渐增加,Pb—Br 键的结合越来越容易,在溶液中不仅会形成[PbBr3]-络合物,还会形成少量的[PbBr4]2-络合物,在纳米晶成核阶段,单斜相CsPbBr3的形核占主导,随着反应的进行,由于TOABr 与Pb2+的持续作用,使得PbBr2不断脱出纳米晶,最后形成CsPbBr3-Cs4PbBr6复合纳米晶,六方相Cs4PbBr6纳米晶的占比会随TOABr 用量的增加而提高.
2)当改变Cs/Pb 摩尔比时,体系中TOABr 的用量为0.32 mmol,过量的TOABr 使得Pb—Br 键更易生成.当Cs/Pb 摩尔比>1 时,体系处于贫铅环境,在前驱体加入早期体系会生成[PbBr3]-和[PbBr4]2-基本骨架,但因为此条件下Pb2+含量较少,大量未被消耗的Br-会使[PbBr3]-向[PbBr4]2-基本骨架转变,使得Cs4PbBr6晶核在成核阶段占据主导,在经历生长阶段后最终获得了六方相Cs4PbBr6纳米晶,导致绿色荧光峰很快消失;当Cs/Pb 摩尔比≤1 时,体系不再具备贫铅环境,在纳米晶成核阶段同时出现[PbBr3]-和[PbBr4]2-基本骨架,两种络合物相互竞争,在形核期,[PbBr3]-占主导,形成了单斜相CsPbBr3NCs,随着PbBr2的不断形成和脱出,单斜相CsPbBr3NCs 会逐渐转变为六方相Cs4PbBr6NCs,使得最终所得纳米晶为CsPbBr3NCs 内嵌入在Cs4PbBr6NCs 中的复合CsPbBr3-Cs4PbBr6NCs.
4 结论
在室温采用单溶剂法在甲苯相合成了相可调的铯铅溴钙钛矿纳米晶,通过调节TOABr 的用量和Cs/Pb 摩尔比系统研究了纳米晶的相结构、形貌、光学性质和结晶/相转变动力学过程.结果表明,当TOABr 浓度为0.16 mmol 时,在反应初期体系中存在大量的[PbBr3]-络合物,促使单斜相CsPbBr3晶核的快速形成,随后晶核经历了尺寸分布聚焦生长和Ostwald 熟化生长后生成尺寸为~12 nm 的CsPbBr3纳米立方块,然而立方块的PLQY 值仅有9%且稳定性较差.随着TOABr 浓度的增大,体系中还会出现[PbBr4]2-络合物,并与[PbBr3]-络合物相互竞争,在反应早期,[PbBr3]-络合物占主导,生成了大量单斜相CsPbBr3纳米晶,随着反应的进行,过量的Br-会与CsPbBr3纳米晶中Pb2+相互作用,使得[PbBr4]2-络合物量逐渐增大,最终形成以CsPbBr3为发光中心的CsPbBr3-Cs4PbBr6复合纳米晶.当TOABr 用量为0.32 mmol时,两相的相对含量分别为58%和42%,PLQY值高达58%,稳定性较好.在此浓度下进一步调节了Cs/Pb 的摩尔比(2∶1,2∶2 和2∶3),结果显示,摩尔比的改变只影响CsPbBr3-Cs4PbBr6复合纳米晶中两相的相对含量,当六方相Cs4PbBr6纳米晶含量较高时,复合材料的稳定性和光学性能较差.
