β-环糊精金属有机骨架材料HPLC手性柱拆分噁唑禾草灵
2024-05-08张家昌陆金富沙涛沈静茹
张家昌,陆金富,沙涛,沈静茹
(中南民族大学 化学与材料科学学院 分析化学国家民委重点实验室,湖北 武汉 430074)
在农业领域,大多数手性农药是作为外消旋混合物施用的,尽管某种农药的生物活性通常只由一种异构体决定,而其他异构体通常用处不大(大部分单一手性源的对象,若能使之分离,即可减少一半的施药量),甚至对非目标生物产生毒副作用,这会误判其潜在的生态健康风险[1]。随着手性农药的概念越来越被接受,目前,学术界已经对其开展了包括手性分离与合成、环境行为、生物代谢、毒理作用等[2]方面的研究,这对手性农药限制的风险评估和监管政策制定至关重要[3]。
噁唑禾草灵(Fenoxaprop-ethyl,FE)是一种芳氧基苯氧丙酸除草剂,研究表明,R-FE是除草活性异构体,FE在生命体中和环境中的代谢物对生物体表现出对映选择性[4]。例如,Xu等[5]研究了FE及其手性代谢物2-(4-羟基苯氧基)丙酸乙酯(EHPP)、2-(4-羟基苯氧基)丙酸(HPPA)和6-氯-2,3-二氢苯并噁唑-2-酮(CDHB)对斑马鱼的影响,研究发现FE、FA、CDHB和EHPP的S型对映体比R型对映体毒性更大。使用FE的单一对映体能有效地降低消旋体施药对生物体和环境的负面影响。目前,分离FE对映体的高效液相色谱(High Performance Liquid Chromatography,HPLC)法,多采用的色谱柱为商业柱。例如,Yan等[6]采用全甲基化β-环糊精商业柱作为固定相,以甲醇和水作为流动相,实现了FE对映体的分离,分析时间大于10 min;Liu等[7]使用Chiralpak IC商业手性柱对R-FE和S-FE的保留时间相差小,且分析时间长。
环糊精金属有机框架材料(Cyclodextrin Metal-Organic Frameworks,CD-MOFs)是理想的手性高效液相色谱柱材料[8]。研究人员可以通过选择不同的金属离子、有机配体和环糊精的类型来调整CD-MOFs的结构和性能。这类MOF材料的多孔结构还可以提高样品与固定相的相互作用,进一步增强分离效率[9]。其能够拆分一些传统手性色谱柱中难以实现的高分子质量化合物,CD-MOFs出色的手性选择性,使之能够有效地分离和鉴定各种手性分子,包括药物、天然产物和农药等[10]。在HPLC手性色谱分析领域,CD-MOFs作为手性色谱柱的应用已经取得了一定的进展[11-12]。尽管如此,这一领域的研究仍然存在一些挑战,如合成方法的进一步改进优化、CD-MOFs的稳定性以及对一些复杂样品的适用性。未来的研究应致力于解决这些问题,并进一步推动CD-MOFs在HPLC手性色谱分析中的应用。
本文采用蒸汽扩散法合成了全新的β-环糊精和间羧基苯磺酰氯金属有机骨架材料,通过加入交联试剂将其键合在全多孔二氧化硅微球表面,形成复合材料(β-CD-MOF-P20@SiO2),以此作为HPLC手性固定相填料。在反相色谱模式下,以乙酸铵-三乙胺/乙酸缓冲液和乙腈作为流动相,对手性农药噁唑禾草灵对映体进行拆分研究,在所探究的色谱条件下,均能实现噁唑禾草灵两对映体的基线分离,分离度RS大于7.30,说明该新型手性固定相对噁唑禾草灵对映体具有好的对映体选择性。在最佳分离条件下,分离度可达9.89,与现有文献报道的工作相比,该体系对噁唑禾草灵对映体分离效果更好,分析速度更快,重现性好,线性范围宽,为噁唑禾草灵对映体的分离分析及单一对映体定性定量和制备提供了新方法。
1 实验部分
1.1 化学试剂和药品
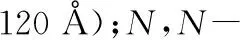
1.2 仪器设备
高效液相色谱仪(HITACHI L-7000,Japan);傅里叶红外光谱仪(Nicolet Nexus 470,USA);紫外-可见-近红外分光光度计(Agilent Cary 5000,USA);X-射线衍射仪(Bruker D8,Germany);场发射电镜(HITACHI SU8010,Japan);真空干燥箱(上海精宏XMTD-8222,中国);集热式恒温加热磁力搅拌器(武汉科尔DF-101S,中国);自动电位滴定计(上海伟业ZD-2型,中国);分析天平(Sartorius ALC-210.4,Germany)。
1.3 手性柱材料的合成
1.3.1β-环糊精掺杂间羧基苯磺酰氯金属有机骨架材料的合成
采用蒸汽扩散法合成:称取适量的β-环糊精、间羧基苯磺酰氯和氯化钾,置于烧杯中搅拌溶解,过滤后转移至密闭容器中,加热反应6 h。反应结束后,自然冷却至室温,过滤,用适量甲醇洗涤数次,烘干,得无色透明晶体产物β-CD-MOF-P20。
1.3.2 手性柱材料β-CD-MOF-P20@SiO2的合成
称取适量上述β-CD-MOF-P20溶于溶剂中,加入一定量的氢化钠,室温下搅拌后将滤液转移至三颈烧瓶中,加入适量偶联剂,在氮气保护下加热反应5 h。然后,加入活化的全多孔二氧化硅微球,冷凝回流反应24 h;反应结束后,冷却至室温,过滤,产物用适量溶剂依次洗涤,放置烘箱中真空干燥,得白色固体粉末。
1.3.3 手性柱填充
称取适量1.3.2的手性柱材料分散在50 mL匀浆液中,将其倒入匀浆罐中,以一定比例洗脱液作为流动相,在25 MPa压力下填充至不锈钢空柱(4.6 mm×150 mm)。
1.4 实验前处理
样品制备:准确称取噁唑禾草灵标准品,以甲醇作为溶剂,配置成标准品原液,逐级稀释成所需样品浓度,并采用微孔滤膜(0.22 μm)过滤备用。
缓冲液制备:准确称取适量乙酸铵,将其溶于超纯水,加三乙胺/乙酸调节缓冲液pH值。
流动相前处理:流动相均采用微孔滤膜(0.22 μm)过滤,并进行超声脱气处理。
2 实验结果与讨论
2.1 材料表征
2.1.1 傅里叶红外光谱表征
使用Nicolet Nexus 470傅里叶红外光谱仪分别对β-环糊精、间羧基苯磺酰氯和β-环糊精掺杂间羧基苯磺酰氯金属有机骨架材料(β-CD-MOF-P20)进行表征,如图1A所示。β-CD-MOF-P20谱图中的3 417 cm-1处的吸收峰为β-CD结构中-OH的伸缩振动吸收,相对于β-CD上-OH的伸缩振动吸收而言,其吸收强度降低,可能是由钾离子与电离的-OH进行配位而导致的;2 923 cm-1处的吸收峰为β-CD结构中-CH的伸缩振动吸收;1 654 cm-1处的吸收峰对应于间羧基苯磺酰氯中C═O的伸缩振动吸收;1 600 cm-1附近的吸收带是由芳环上C═C的伸缩振动引起的,β-CD-MOF-P20的谱图同时包含有β-环糊精和间羧基苯磺酰氯的特征吸收峰,部分吸收峰发生偏移,初步说明β-CD-MOF-P20的成功合成。
(a)傅里叶红外光谱图;(B)紫外-可见吸收光谱图;(C)X-射线粉末衍射谱图;(a)β-环糊精;(b)间羧基苯磺酰氯;(c)β-CD-MOF-P20。
2.1.2 紫外可见吸收光谱表征
采用Agilent Cary 5000紫外-可见-近红外分光光度计分别测定了2.1.1中三种材料的固体紫外可见吸收光谱,结果如图1B所示。间羧基苯磺酰氯在218和284 nm处有吸收峰,分别对应苯环的K吸收带和E吸收带;β-CD-MOF-P20的光谱图显示,在233和274 nm出现两处吸收峰,应是由间羧基苯磺酰氯引入导致的,间羧基苯磺酰氯中苯环的K和E吸收带引起了吸收峰吸收强度的变化且发生了位移,说明β-环糊精和间羧基苯磺酰氯共同参与了β-CD-MOF-P20材料的合成。
2.1.3 X-射线粉末衍射表征
采用HITACHI SU8010 X-射线衍射仪分别对上述三种材料进行表征,如图1C所示。从图中信息可以看出,β-CD-MOF-P20在6.7°,9.6°,11.0°,12.9°,13.5°,18.3°处有明显衍射峰,β-环糊精中包含相近的衍射峰,β-CD-MOF-P20的X-射线衍射谱图与β-环糊精、间羧基苯磺酰氯有不同之处,衍射峰大于20°后其未见明显吸收峰,说明β-CD-MOF-P20形成了新的晶型,进一步说明β-CD-MOF-P20的成功合成。
2.1.4 扫描电子显微镜表征
采用Bruker D8场发射电镜分别对二氧化硅微球、β-CD-MOF-P20和β-CD-MOF-P20@SiO2进行表征,如图2所示,其中图2a为二氧化硅微球裸珠,表面光滑;图2b为β-CD-MOF-P20@SiO2,与二氧化硅微球裸珠相比,表面粗糙,表面颗粒粒径变大且生长均匀,说明β-CD-MOF-P20是成功生长在二氧化硅微球表面的MOF材料。

(a)SiO2裸珠;(b)β-CD-MOF-P20@SiO2。
2.2 分离噁唑禾草灵对映体的色谱条件探究
选择色谱条件:柱温箱温度T=25 ℃;检测波长λ=254 nm;进样体积V=20 μL。
2.2.1 流动相配比的影响
以乙酸铵-乙酸(c=20 mmol/L,pH值4.40)和乙腈作为流动相,通过改变流动相配比,探究流动相配比对FE对映体分离的影响,实验结果如图3(a)所示。随着乙腈占比的下降,R-FE峰型逐渐尖锐,峰高升高,保留时间变化不大;而S-FE峰型逐渐展宽,峰高降低,保留时间增加;分离度逐渐升高,即缓冲液占比的上升有利于FE对映体的分离。手性色谱柱选择性地保留S-FE,说明缓冲液有利于FE进入β-CD衍生物空腔,形成包埋复合物,而β-CD衍生物与S-FE形成的包埋复合物更加稳定,进而影响S-FE的洗脱。
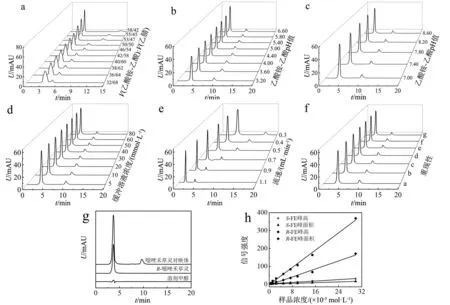
(a)流动相不同比例;(b)乙酸铵-乙酸不同pH值;(c)乙酸-三乙胺不同pH值;(d)缓冲液不同浓度;(e)流动相不同流速;(f)重现性试验;(g)对照实验;(h)线性相关性。
2.2.2 缓冲液pH值的影响
综合考虑,选定缓冲液和乙腈的体积比为53∶47作为流动相配比,其他色谱条件不变,通过改变缓冲液pH值,探究缓冲液pH值对FE对映体分离的影响,实验结果如图3(b)、(c)所示。缓冲液pH值范围从酸性到碱性,分析物的保留时间、峰型和分离度均没有明显变化,说明在该缓冲液中,R,S-FE对映体分离在较宽的pH值范围均能被拆分,pH值影响不大,可能是在此环境下FE的结构相对稳定,改变缓冲液pH值并未使其有大的变化。
2.2.3 缓冲液浓度的影响
选定缓冲液pH值为7.40,其他色谱条件不变,通过改变缓冲液浓度,探究缓冲液浓度对FE对映体分离的影响,实验结果如图3(d)所示。随着缓冲液浓度的升高,除了S-FE的保留时间呈下降趋势外,其他前后峰的参数均未出现明显的变化,结果导致分离度呈下降趋势,当缓冲液浓度为20 mmol/L时,分离度最佳,RS达9.09。实验结果说明,缓冲液浓度在一定程度上影响了S-FE与β-CD衍生物之间形成的非对映体复合物的稳定性。
2.2.4 流动相流速的影响
选定缓冲液浓度为20 mmol/L,其他色谱条件不变,通过改变流动相流速,探究其对FE对映体分离的影响,实验结果如图3(e)所示。随着流动相流速的升高,前后峰的保留时间和峰面积逐渐减小,峰型逐渐尖锐,分离度先升后降,当流速为0.5 mL/min时,分离度最佳,RS达9.09。
2.2.5 对照实验
在流动相乙酸铵-三乙胺(20 mmol/L,pH值7.40)和乙腈配比为53∶47(体积比),温度25 ℃,检测波长254 nm,流速为0.5 mL/min的色谱条件下,分别测定了溶剂甲醇、R-FE和FE消旋体样品,实验结果如图3(g)所示。通过对照实验说明,溶剂甲醇对分离实验结果的干扰不大,前峰对应的对映体为R-FE,则后峰对应的对映体为S-FE。
2.2.6 重现性试验
以2.2.5同样的色谱条件下,依次进样7次,通过确定相对标准偏差(RSD)来评估该体系的重现性,实验结果如图3(f)所示。R-FE的保留时间、峰高、峰面积的RSD值分别为0.09%,0.77%和0.68%,S-FE的保留时间、峰高、峰面积的RSD值分别0.59%,1.32%,1.53%,分离度的RSD值为0.91%。实验结果表明,该体系分离FE对映体具有良好的稳定性和重现性。
2.2.7 线性相关性
以2.2.5同样的色谱条件下,在FE消旋体标准品浓度在6.20×10-6~2.99×10-4mol/L范围内分别分离了8个不同浓度的试样,依次进样,将FE两个对映体的峰高和峰面积分别与样品浓度进行线性拟合,实验结果如图3(h)所示。S-FE的峰高和峰面积与样品浓度的线性方程分别为:y=1.255 9x-0.305 5(r=0.996 2)和y=0.481 7x+0.082 9(r=0.999 6),R-FE的峰高和峰面积与样品浓度的线性方程分别为:y=11.563 9x+0.122 1(r=0.996 7)和y=4.670 2x-0.538 9(r=0.999 9)。结果表明,FE两个对映体的峰高和峰面积与样品浓度(6.20×10-6~2.99×10-4mol/L)线性相关性好(r=0.996 2~0.999 9),线性范围宽。
3 结论
采用以β-CD衍生物为手性源的CD-MOF手性HPLC固定相,开发了一种分离R,S-FE对映体的反相色谱方法,通过优化色谱条件,得到了流动相乙酸铵-三乙胺(20 mmol/L,pH值7.40)和乙腈配比为53∶47(体积比),温度25 ℃,检测波长254 nm,流速0.5 mL/min的色谱条件下获得了好的FE对映体分离,与已有报道的比,该材料对R,S-FE对映体的分离具有更大的分离度和更快速的优势,该工作对更好地评估FE手性农药的环境和生态风险具有一定意义,也会促进相关研究的发展。
