基于虚拟筛选从中药中发现高选择性靶向DDX3X抑制剂
2024-04-30史学伟王宝珍董志强
邵 晨 宋 昱 史学伟 王宝珍 武 靖 董志强*
人类免疫缺陷病毒1型(human immunodeficiency virus type 1, HIV-1)属于逆转录病毒科,是全球公共卫生领域面临的最严峻挑战之一,对人类健康构成极大威胁。尽管1996年开始引入抗逆转录病毒疗法,但据估计,全世界仍有3 670万HIV-1携带者,每年死亡人数超过100万[1-2]。由于HIV-1潜伏期长且易变异,迄今为止彻底根除这种病原体或开发有效的预防性或治疗性疫苗相当困难[3]。随着医学技术发展,现有抗病毒药物的效力提高,HIV-1的复制可以在大多数接受治疗的患者中成功地停止[4]。此外,免疫功能低下的患者易受细菌、真菌和病毒感染,这使得治疗方案变得复杂,增加了用药负担[5]。
为了改善药物选择性并解决耐药性问题,间接作用药物被提出。HIV-1等病毒在宿主细胞内复制时,需借助宿主细胞的各类分子和生化途径。这些由宿主细胞提供的辅助因子成为了抑制病毒复制的潜在药物靶点[6]。DEAD-box家族包含大量已知被不同病毒吸收的人类蛋白质,许多不同的病毒依赖其调节宿主细胞的反应,DEAD-box家族中的ATPase/RNA helicase X-linked DEAD-box polypeptide 3(DDX3X)蛋白被确定为是许多不同的病毒调节宿主细胞的依赖蛋白[7]。近年来,针对DDX3X的ATP和RNA结合位点开发出许多小分子抑制剂,尤其是发现了一种针对DDX3X的核酸结合裂缝的RNA竞争抑制剂,它能够阻断HIV-1以及丙型肝炎病毒(hepatitis C virus, HCV)、登革病毒(dengue virus, DENV)和西尼罗河病毒(west nile virus, WNV)的复制[8-10]。同时DDX3X参与不同的细胞途径(翻译、转录、RNA衰变、核糖体生物发生),参与细胞周期调控、凋亡、癌变、迁移和缺氧,并在大量肿瘤细胞中过度表达,被认为是抗癌治疗学发展的新靶点[11-12]。
然而,大多数DEAD-box家族中RNA解旋酶的ATP和RNA结合位点具有显著的相似性,这对开发具有绝对选择性的药物带来了一定的挑战,除了所有典型的保守结构域外,DDX3X还被证明在参与ATP结合的基序I(WalkerA)和属于RNA结合域的基序Ia之间有一段独特的序列(ALRAMKENGRYGRRK,250~264),该序列被称为“独特的基序”[13-14]。值得注意的是,这段序列在DEAD-box家族中的其他蛋白成员中并不存在,而且文献[15]报道表明,删除这个短基序会降低DDX3X与核酸的亲和力,从而影响DDX3X的RNA解旋酶活性,而用肽封闭这个结构域可以减少感染细胞中的HIV-1复制。因此,靶向这一独特基序的抑制剂对DDX3X具有高度的选择性,并可能保留抑制病毒复制的能力。
在本实验中,针对这一独特的位点基于中药化合物数据库筛选出选择性较强的化合物,通过分子对接、互作模式分析以及计算自由结合能的方法筛选出具有潜在抑制活力的DDX3X抑制剂,并对活性位点的构效关系进行分析,为DDX3X高选择性抑制剂设计提供指导方案。
1 资料与方法
本实验所有分子模拟工作运用Schrӧdinger 2019软件中各模组完成,Discovery Studio 2020用于视图操作。
1.1 蛋白准备
从PDB数据库(https://www.rcsb.org)下载DDX3X蛋白晶体结构(PDB ID:2I4I)[16],使用Schrӧdinger将蛋白晶体结构中的水分子删掉,在“Protein preparation wizard”模块中,对蛋白进行加氢以及对蛋白晶体结构中非完整氨基酸残基进行矫正。同时,根据PROPKA的pKa预测,在pH为7.0下实现质子化残基的氢键优化。最后,在OPLS3立场下使蛋白晶体结构能量最小化。
1.2 小分子准备
模拟过程中的全部小分子均利用Schrӧdinger中“LigPrep”模组进行处理,将小分子在OPLS3立场下进行结构优化,不考虑异变体的因素下,每个小分子最多只生成一个立体异构体。
1.3 活性位点确定
识别DDX3X上ATP结合的基序I(WalkerA)和属于RNA结合域的基序Ia之间独特的序列(ALRAMKENGRYGRRK, 250~264),将该序列内氨基酸构成的口袋定义为选择性位点,基于此位点进行选择性抑制剂筛选。利用SiteMap模块对基序进行识别,将识别后产生的位点定义为选择性位点。
1.4 分子对接
模拟实验中分子对接部分由Schrӧdinger中“Glide”模组完成。通过对活性位点的确定,在选择性位点中利用Glide中的HTVS对接完成初期的对接筛选,随后SP对接进一步确定配体-蛋白的结合效果。对接精度较高的XP对接用来分析筛选得到的化合物与蛋白的互作模式。对接实验前要评价对接方法的可靠性,将共晶体中的共晶化合物AMP进行重新对接,分析对接前后的RMSD值,RMSD值小于2Å认为对接结果具有可靠性。
1.5 自由结合能计算
自由结合能可反映配体-蛋白复合物体系的稳定性,通过Prime MM-GBSA对复合物体系自由结合能进行计算。自由结合能计算遵循以下方程:
ΔG bind=ΔGMM+ΔG solv+ΔG SA
ΔG bind的计算涉及到3个主要方面:气相自由能(ΔGMM)、溶剂化自由能(ΔG solv)和系统熵变化(ΔG SA)。为了实现这一计算,本研究选择溶剂模型为VSGB,并采用“层次采样”以高效而系统的方式对配体-蛋白复合物进行位置、方向和构象的系统采样,以全面捕捉其结合过程中的各种可能性。同时,在OPLS3力场下,对各个配体-蛋白复合物体系自由结合能进行计算。
2 结果
2.1 选择性位点分析
目前,大多数DDX3X蛋白解旋酶抑制剂主要瞄准其RNA结合位点,然而,特定位点(250~264)的独特序列在其他家族蛋白中缺失,为增强对DDX3X的选择性提供了潜在优势。利用SiteMap程序对“独特的基序”进行识别。如图1所示,定义选择位点的中心为X=27.6,Y=0.16,Z=5.02。选择性位点位于蛋白末端,由一小段α螺旋构成,螺旋末端连接着一段loop环。对选择性位点进行分析,位点内的氨基酸多为亲水性氨基酸,这些氨基酸具有与小分子抑制剂形成氢键的潜力,此外多数氨基酸残基在口袋内呈正电性或负电性。
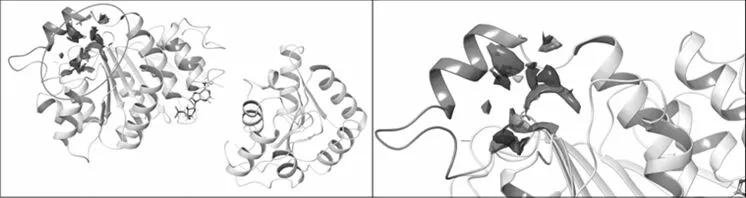
图1 选择性位点预测
2.2 分子对接筛选
分子对接实验前,对分子对接方法进行了评价。采用配体扩张法定义共晶化合物(X=17.2,Y=-23.33,Z=16.36)10Å范围内的氨基酸为活性位点,将共晶化合物AMP重新对接后计算对接前后AMP构象的变化。测得RMSD值为0.293Å,表明本实验分子对接方法具有一定的可靠性。首先基于高通量分子对接筛选方法对化合物数据库中15万个小分子进行筛选,结果显示共有123 199个化合物可以结合在选择性位点。随后调整对接精度为SP,将这些化合物与选择性位点再次对接,随后对结合分数进行统计,选取结合分数排名前3 000的化合物进一步进行精确度最高的XP对接。设置对接方式为半柔性对接,将这些候选化合物与选择性位点对接。观察分析对接后化合物的分子构象,剔除那些与选择性位点构象不符合的化合物,同时对化合物的结合分数进行分析,最终确定了20个化合物有着可以与选择性位点相匹配的分子构象,同时有着较好的结合作用。20个化合物理化性质如表1所示。其中化合物1在这些化合物中表现出较强的作用效果,如图2所示,化合物1分子构象由3个环状结构构成,三部分结构均匀地分布在选择性位点中,且3个环状结构的羟基均与活性位点中的氨基酸形成氢键。

表1 化合物理化性质
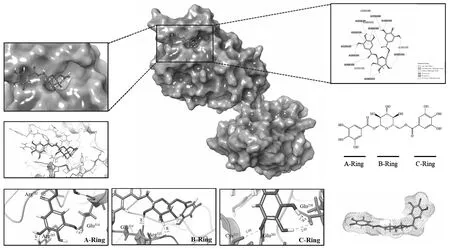
图2 化合物1于选择性位点作用方式
2.3 自由结合能计算
分子对接筛选得到的化合物表现出可与DDX3X蛋白解旋酶形成较强的结合作用,但单纯的分子对接实验并不能证明配体与蛋白可构成稳定的复合物体系,需通过MM-GBSA对分子对接筛选出的化合物与DDX3X蛋白组成的复合物体系自由结合能进行计算,分析蛋白-配体复合物体系的稳定性。在进行自由结合能计算时,设置化合物3Å范围内的氨基酸为柔性构象。20个化合物的自由结合能如表2所示,以ΔGbind=-60 kcal/mol为标准,选择自由结合能低于该数值的化合物,最终确定了11个化合物。XP精准对接结果显示化合物1与DDX3D蛋白解旋酶有着较好的结合效果,自由结合能计算结果显示两者构成的复合物体系较为稳定,其中库伦能(ΔGbindCoulomb=-70.239 kcal/mol)以及范德华作用(ΔGbindvdW=-45.423 kcal/mol)提供了主要贡献。库伦能表现为静电作用,选择性位点上氨基酸Arg、Lys、Asp以及Glu在对接环境下,氨基酸上的羟基与氨基电离形成带电单位,如图2所示化合物分子表面电势图显示出较强的负电性,化合物1不同部位的负电性与选择性位点上带正电荷氨基酸Lys以及Arg结合形成较强的静电作用以及氢键(ΔGbindHbond=-7.702 kcal/mol)作用,从而提升复合物的稳定性。其他化合物与化合物1相似,库仑能以及范德华作用为主要贡献者,其次是氢键以及疏水作用。自由结合能计算结果与选择性位点特征相符,即位点上以带电荷氨基酸为主,与这些氨基酸发生静电作用和氢键作用可增强化合物在选择性位点的结合效果。
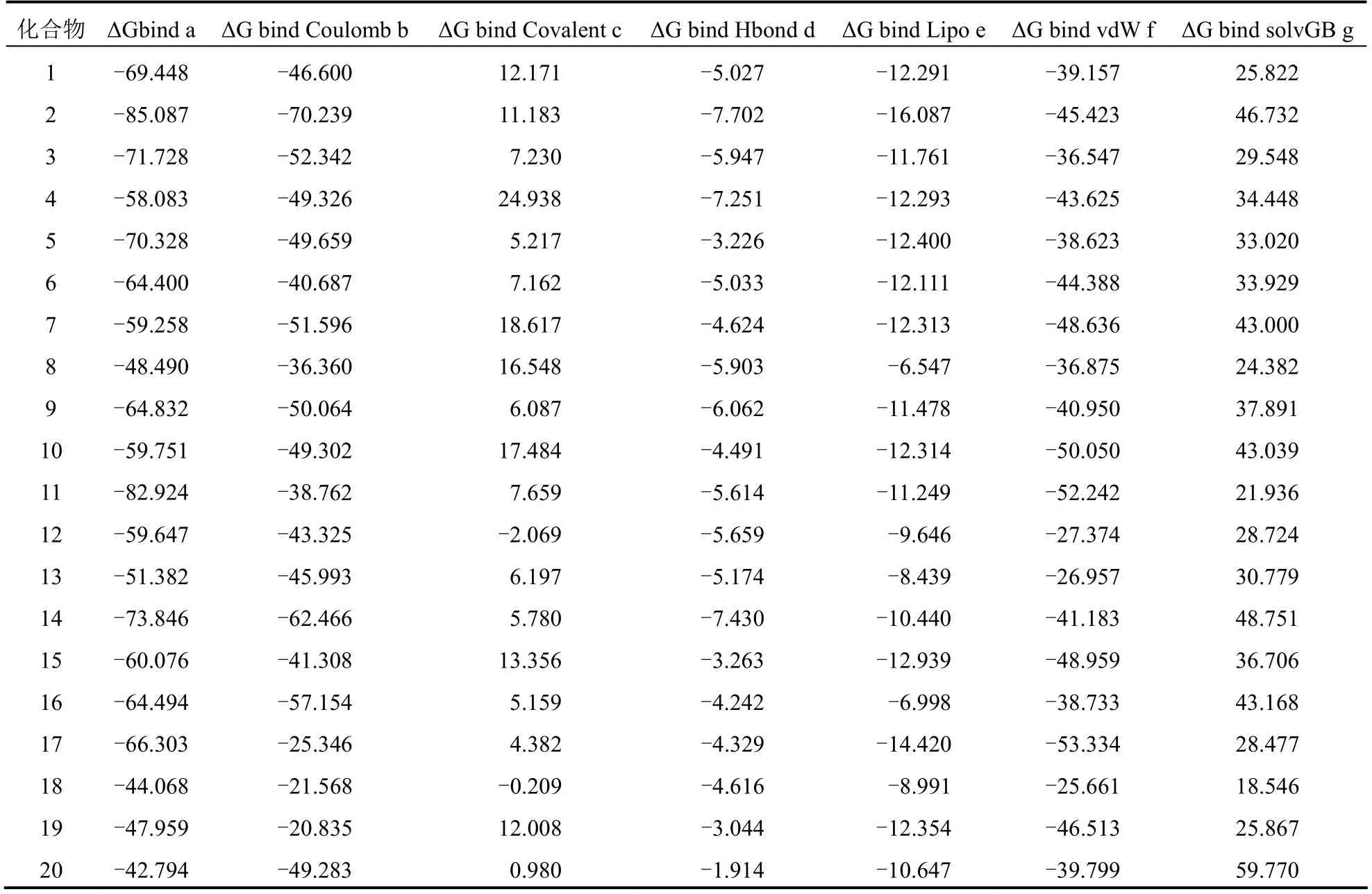
表2 蛋白-配体自由结合能(kcal/mol)
2.4 互作模式
配体-蛋白间的作用模式决定了复合物体系的稳定性,与关键氨基酸存在相互作用可增强化合物对蛋白的作用效果。通过DS可视化工具对这11个化合物与DDX3X蛋白的作用方式进行精确分析。如表3所示,氢键和范德华力是11个化合物中保守的两种作用力。氨基酸Glu249、Glu256、Arg262、Gln265以及Arg315可以与多个化合物形成氢键作用,此外化合物1、2、5、6、14和15还可以与Glu256以及Arg262带正电荷氨基酸形成静电作用,这进一步加强了这些化合物与DDX3X蛋白解旋酶的结合效果。氨基酸Arg296、Arg315以及Gly316与大部分化合物形成范德华力作用。除氢键与范德华力这两种保守作用力外,化合物2、9、11以及14与氨基酸Arg296、Arg252、His318等残基上的疏水基团存在疏水作用。见图3~4。

表3 配体-蛋白作用方式
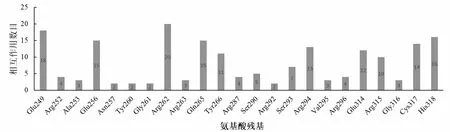
图3 XP对接后打分TOP20的分子与受体各氨基酸残基产生相互作用的数目

图4 结合能排名前三的化合物与DDX3X蛋白解旋酶选择性位点作用方式
通过对11个化合物与蛋白的互作模式进行分析,结合自由结合能计算结果,表明在选择性位点中,与位点上的氨基酸形成氢键的同时存在静电作用可进一步加强化合物与DDX3X蛋白解旋酶的稳定性。氨基酸Glu249、Glu256、Arg262、Gln265以及Arg315作为关键氨基酸与化合物形成氢键以及静电相互作用。通过对化合物与DDX3X蛋白解旋酶互作模式分析,认为化合物1、2、5以及14相比于其他化合物可稳定地作用于DDX3X蛋白解旋酶选择性位点并与蛋白形成更加稳定的复合物体系。
3 讨论
本实验基于DDX3X蛋白解旋酶上一段独特的基序构建选择性位点,针对该选择性位点对中药数据库进行虚拟筛选,筛选分析得到强效DDX3X蛋白解旋酶选择性抑制剂。通过分子对接、自由结合能计算以及互作模式分析的方法确定选择性位点以亲水性氨基酸为主且多数为带正电荷氨基酸。化合物1、2、5和14(ZINC4096316、ZINC33861462、ZINC67903526、ZINC85530944)可与选择性位点内的Glu249、Glu256、Arg262、Gln265以及Arg315关键氨基酸形成氢键以及静电作用,与氨基酸Arg296、Arg315以及Gly316等形成范德华力。两种保守的作用力使得这4种化合物可以与DDX3X蛋白解旋酶形成稳定的复合物体系,4种化合物可作为潜在的高选择性DDX3X蛋白解旋酶抑制剂进一步开发。
