UPLC-MS/MS测定猪肉中头孢类药物的残留量
2024-04-29孙钰莹李启卉陈月
摘 要:本文采用超高效液相色谱-串联质谱对猪肉中头孢类药物残留量进行分析。样品经乙腈-水溶液和磷酸盐缓冲溶液提取,HLB固相萃取柱纯化后用超高效液相色谱质谱联用仪进行测定。结果表明,3种头孢类药物浓度在0~200 ng·mL-1时,相关系数大于0.996,线性关系良好;检出限为0.001~0.365 μg·kg-1,定量限为0.003~1.220 μg·kg-1。不同浓度下样品的加标回收率为83.2%~95.2%,相对标准偏差为1.9%~7.5%,标准偏差为0.078~2.817 μg·kg-1。
关键词:猪肉;头孢类药物;超高效液相色谱-串联质谱;固相萃取柱
Determination of Cephalosporin Residues in Pork by UPLC-MS/MS
SUN Yuying, LI Qihui, CHEN Yue
(National Light Industry Food Quality Supervision and Testing of the Nanjing Station ,Nanjing 211816, China)
Abstract: This article uses ultra high performance liquid chromatography tandem mass spectrometry to analyze the residual levels of cephalosporins in pork. The sample was extracted by acetonitrile aqueous solution and phosphate buffer solution, purified by HLB solid-phase extraction column, and determined by ultra-high performance liquid chromatography mass spectrometry. The results showed that the correlation coefficient (R2) of the three cephalosporin drugs was greater than 0.996 within 0~200 ng·mL-1, indicating a good linear relationship; the detection limit is 0.001~0.365 μg·kg-1, with a quantification limit is 0.003~1.220 μg·kg-1. The spiked recovery rates of samples at different concentrations is 83.2%~95.2%, with a relative standard deviation is 1.9%~7.5% and a standard deviation is 0.078~2.817 μg·kg-1.
Keywords: pork; cephalosporins; ultra performance liquid chromatography tandem mass spectrometry; solid-phase extraction column
兽药可作为生长促进剂,在畜牧业中还被广泛用于疾病的治疗和预防。但兽药使用不当会导致动物源性食品中存在兽药残留物。头孢菌素类抗生素由天然头孢菌素C衍生而来,被广泛用于治疗和预防细菌感染的疾病,如牛乳腺炎、腹泻病毒等。由于其具有良好的治疗效果、低致敏性、无毒副作用等优点,是目前最常用的抗菌药物。但头孢菌素类药物的不当使用可能会使肉制品中残留该药物并危害人们的健康,如耐药性和过敏反应。目前对头孢类药物残留分析的方法主要有高效液相色谱法、毛细管电泳法、液相色谱-串联质谱法和微生物法,由于上述方法通常烦琐且缺乏选择性,而超高效液相色谱-串联质谱(Ultra Performance Liquid Chromatography Tandem Mass Spectrometry,UPLC-MS/MS)具有高特异性、高灵敏性、高准确性等优势[1],本研究采用UPLC-MS/MS方法对猪肉中3种头孢类药物的残留量进行分析,为测定猪肉中头孢类药物残留量提供一种新思路。
1 材料与方法
1.1 材料与试剂
猪肉,购自盒马;头孢类药物标准品(纯度均大于95%,上海安谱璀世标准技术服务有限公司);乙腈(质谱纯,默克);甲酸(色谱纯);磷酸二氢钾(分析纯);Oasis PRiME HLB固相萃取小柱(200 mg/6 mL)。
1.2 仪器与设备
Waters-TQ-Cronos液相色谱质谱联用仪;BSA124S-CW电子天平(万分之一);移液器(艾本德中国有限公司);离心机(上海安亭-GL-20G-Ⅱ);涡旋仪(其林贝尔仪器-Vortex QL-861);BFAA-DC24-RT加热氮吹仪(上海安谱实验科技股份有限公司)。
1.3 方法
1.3.1 样品前处理
用10 mL乙腈-水溶液提取2 g猪肉样品,均质提取1 min,10 000 r·min-1离心5 min,上清液转移至鸡心瓶中,重复提取1次后将上清液合并至鸡心瓶中。40 ℃旋转蒸发除去乙腈,立即加入20 mL磷酸盐缓冲溶液,涡旋混匀后待净化。移取上清液至HLB固相萃取小柱中,依次用3 mL磷酸盐缓冲溶液,2 mL水淋洗,5 mL甲醇洗脱于离心管中。将提取的溶液氮吹蒸发至干,然后用1mL 0.1%甲酸水-乙腈溶液溶解残留物,涡旋混匀,经0.22 μm滤膜过滤后,供超高效液相色谱串联质谱进行测定分析。
1.3.2 标准曲线配制
分别移取头孢氨苄、头孢匹林、头孢唑啉标准储备液250 μL于10 mL容量瓶中,用乙腈-水溶液稀释至刻度线,配制成浓度为10 μg·mL的混合标准中间液。吸取100 μL混合标准中间液,用乙腈-水稀释至1 mL,配制成浓度为1 μg·mL-1的标准使用液。标准曲线的浓度范围为0~200 ng·mL-1,配制完成后上机测定。
1.3.3 色谱条件
色谱柱为ACQUITY UPLC®HSS T3(2.1 mm×100 mm,1.8 μm);柱温30 ℃,流动相A为0.1%甲酸水,B为乙腈;流速为0.25 mL·min-1;进样量5 μL。
1.3.4 质谱条件
ESI正离子模式;监测模式:MRM;雾化器温度:500 ℃;雾化器流速:800 L·h-1;毛细管电压:0.50 kV。
2 结果与分析
2.1 色谱条件
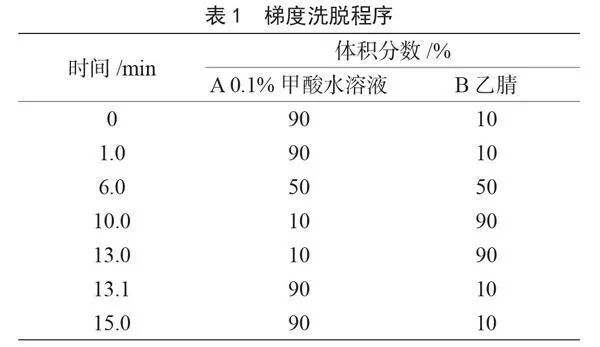
流动相A相为0.1%甲酸水,B相为纯乙腈,采用梯度洗脱时,检测物的峰型良好,具有较高的灵敏度。在正电离模式下,甲酸可以提供丰富的质子供应,有利于[M+H]+离子的形成,使目标物之间、目标物与溶剂峰之间能够较好分离[2],因此本研究选择0.1%甲酸水-乙腈作为流动相。本实验中3种头孢类药物的保留时间为2.0~6.0 min,调整有机相比例和梯度洗脱,切换阀控制主峰不流入质谱,同时确保每次样品测定时主峰能被完全洗脱而不影响下一针样品的检测,梯度洗脱程序见表1。
2.2 质谱条件
分别配制3种头孢类药物的标准溶液
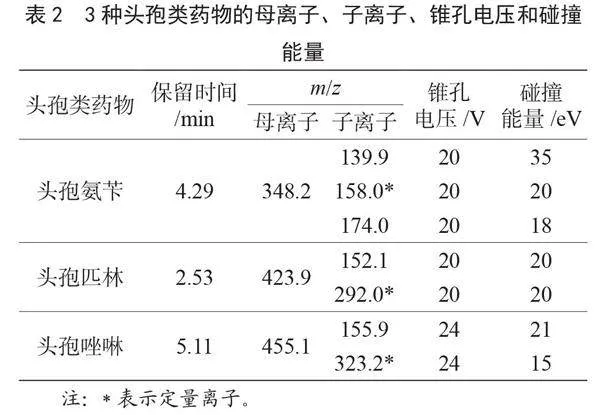
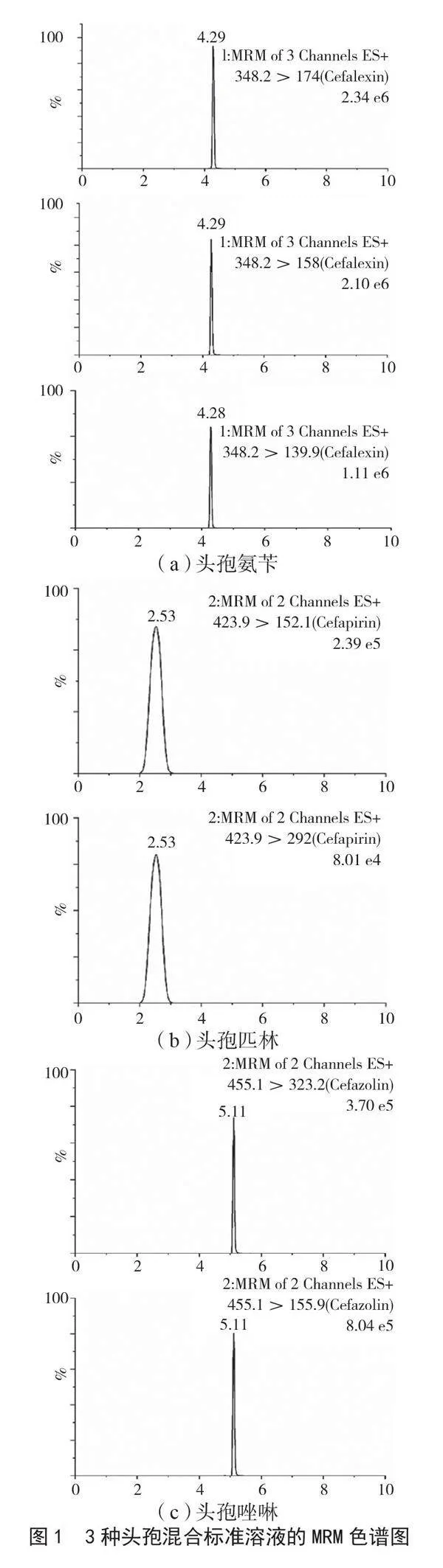
(200 ng·mL-1),采用ESI+模式扫描,选择相对丰度较高、出峰稳定的母离子和子离子碎片。母离子和子离子碎片通过ESI柱洗脱液进入施加电压的不锈钢毛细管中,加热的气体(通常为氮气)沿着毛细管传递,导致洗脱液雾化和带电液滴产生,而这些液滴在经历过库仑裂变,进入质谱仪前便通过解吸和解溶转化为稳定的离子[3]。3种头孢类药物最优的UPLC-MS/MS检测参数如表2所示。其标准溶液MRM色谱图见图1。各药物的色谱峰峰型尖锐且保留时间处无杂峰干扰。
2.3 固相萃取小柱的选择
HLB固相萃取小柱对疏水化合物或亲水化合物都有良好的适应性[4-5],能有效保留甘油酯、磷脂、蛋白质等化合物,且使用前无须活化和平衡,可有效富集净化目标成分,提高检测的准确度,综合实验对比,本实验用HLB作为固相萃取小柱。
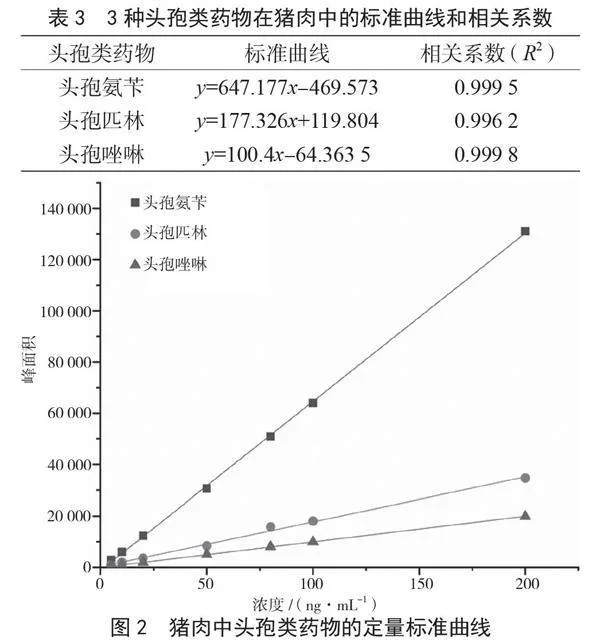
2.4 标准曲线的绘制
采用外标定量法,绘制猪肉中头孢类药物的定量标准曲线,并求出回归方程和相关系数(R2),得到的线性回归方程见表3,标准工作曲线如图2所示。当3种头孢类药物的浓度在0~200 ng·mL-1时,线性关系良好,相关系数R2为0.996 2~0.999 8,均大于0.99。
2.5 回收率、精密度与检出限测定结果
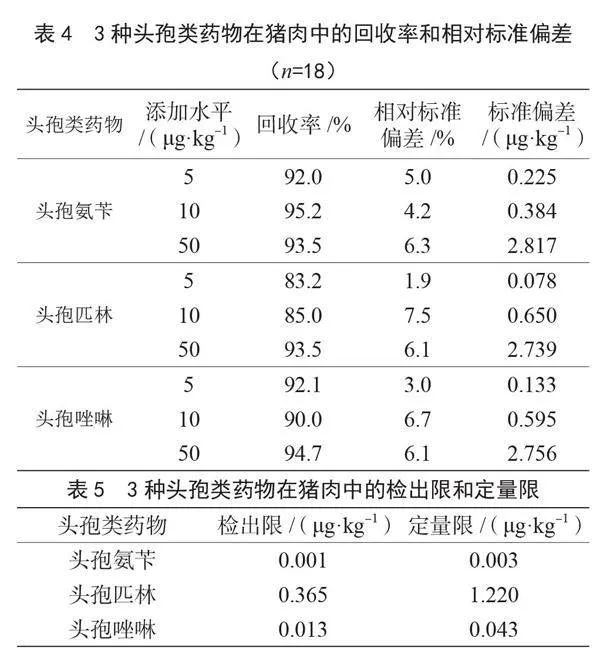
在猪肉空白基质中进行加标回收实验,添加水平为5 μg·kg-1、10 μg·kg-1、50 μg·kg-1共3个浓度,每批次添加水平做6个平行样品,重复测定3次。由表4可知,3种头孢类药物的加标回收率为83.2%~95.2%,相对标准偏差为1.9%~7.5%,标准偏差为0.078~2.817 μg·kg-1。结果表明,该方法具有较好的精密度和准确度。
分别以3倍信噪比和10倍信噪比时添加的质量浓度作为方法的检出限和定量限,结果如表5所示。3种头孢类药物的检出限为0.001~0.365 μg·kg-1,定量限为0.003~1.220 μg·kg-1。根据《出口动物源食品中头孢类抗生素残留量的测定 液相色谱-质谱/质谱法》(SN/T 1988—2023)规定头孢氨苄、头孢匹林、头孢唑啉在动物源食品中的检出限为2.0 μg·kg-1,定量限为5.0 μg·kg-1,而本方法检出限为0.001~0.365 μg·kg-1,定量限为0.003~1.220 μg·kg-1,能满足头孢类药物含量分析的需要。
3 结论
本实验建立UPLC-MS/MS法同时测定猪肉中头孢氨苄、头孢匹林、头孢唑啉的含量,结果表明3种头孢类药物在0~200 ng·mL-1相关系数(R2)大于0.996,线性关系良好;其中方法检出限为0.001~0.365 μg·kg-1,定量限为0.003~1.220 μg·kg-1。样品的加标回收率为83.2%~95.2%,相对标准偏差为1.9%~7.5%,标准偏差为0.078~2.817 μg·kg-1。建立的方法快速、简单,重复性和选择性好,适用于猪肉样品中头孢类药物残留量的检测。
参考文献
[1]薛雨,陈宇瑛.头孢菌素类抗生素的最新研究进展[J].中国抗生素杂志,2011,36(2):86-92.
[2]FENG Q,ZHOU J,ZHANG Y.Coupling Bi2MoO6 with persulfate for photocatalytic oxidation of tetracycline hydrochloride under visible light[J].Journal of Materials Science:Materials in Electronics,2019,30(21):19108-19118.
[3]胡秀虹,张廷辉,王翔,等.微乳法制备纳米N-TiO2及其光降解头孢氨苄性能[J].化学研究,2019,30(2):170-175.
[4]张晶,赵兴然,张群,等.HPLC法同时测定动物性食品中5种头孢类抗生素[J].食品工业,2016,37(2):269-272.
[5]李敏,乔卫叶,王秀玲,等.离子液体为电解质背景毛细管电泳法同时测定四种注射用头孢药物的含量[J].化学研究与应用,2017,29(5):604-609.
作者简介:孙钰莹(1997—),女,福建福州人,硕士。研究方向:食品检测。
