转录组结合区域关联分析挖掘油菜含油量积累的候选基因
2024-04-28向星汝刘忠松官春云钱论文熊兴华
曹 松 姚 敏 任 睿 贾 元 向星汝 李 文 何 昕 刘忠松 官春云 钱论文 熊兴华
转录组结合区域关联分析挖掘油菜含油量积累的候选基因
曹 松 姚 敏 任 睿 贾 元 向星汝 李 文 何 昕 刘忠松 官春云 钱论文*熊兴华*
湖南农业大学农学院, 湖南长沙 410128
油菜(L.)是中国食用植物油的主要来源, 提高种子含油量是增加菜籽油供应最为有效的方法。本研究利用4个油菜自交系授粉后25 d、35 d、45 d的种子转录组数据分析, 筛选出43个与油脂合成相关基因, 其中33个基因持续上调表达, 10个基因持续下调表达, 主要基因包括、、和等。同时, 结合50份半冬性甘蓝型油菜重测序数据, 检测到与含油量显著相关3个SNP、9个SNP分别定位到A10和A05, 其中-A10_Hap1对应材料含油量显著高于Hap2,A05_Hap1对应材料含油量显著高于Hap3。此外, 利用WGCNA构建基因共表达网络发现,与通过3个转录因子LEC1、HMGB3、HTA11间接相连, 形成了潜在调控的分子网络, 影响种子油脂积累。这些结果有利于我们开发单体型功能标记进一步提高油菜籽含油量。
甘蓝型油菜; 含油量; 转录组分析; 共表达分析; 区域关联分析
油菜是异源四倍体油料植物(AACC, 2=38), 大约在7500年前由2个二倍体植物(AA, 2=20)和(CC, 2=18)杂交, 通过自然加倍而成。在全球范围内, 它是植物油的第三大来源, 约占人类消费植物油总量的15%。油菜含油量一般在35%~50%之间[1], 极少数品种的含油量能够达到60%以上[2]。王汉中[3]研究表明, 含油量增加1%, 相当于种子产量增加了2.3%。因此, 提高油菜含油量对油菜产业的发展具有重要意义。
油菜的含油量主要受母体效应、胚胎基因效应、花粉直感、细胞质效应以及相应的基因-环境相互作用效应控制, 符合加-显-上位遗传模型, 基于加性和显性遗传, 具有较高的广义遗传力[4-6]。含油量在种子发育过程中具有明显的动态变化, 与植物光合作用、种子发育、油脂合成转运、油脂积累降解等多种生物途径密切相关, 形成了多基因调控网络[7-9]。种子油脂生物合成分为脂肪酸合成和三酰基甘油合成2个阶段, 主要以三酰基甘油的形式储存, 占种子质量的60%[10]。三酰基甘油的合成需要许多亚细胞结构和多种途径的相互作用, 并且整个过程涉及许多酶基因[11-12]。关键酶编码基因在油菜油脂的合成中起着重要作用, 其对油菜SOC的影响主要取决于基因的表达和酶活性的调控, 参与这一过程的基因主要催化脂肪酸链和三酰基甘油的合成, 包括、、、、和[13-15], 任何一种酶的表达或调控都会影响SOC。关键转录因子也会影响油菜中的SOC, 主要调节种子发育和种子油脂积累。有研究表明, 包括WRI1、LEC1、LEC2、FUS3和ABI3在内的转录因子可以通过激活或抑制与种子油脂合成相关的基因的表达来增加种子油含量[16-20]。此外, miRNA也可能参与油脂代谢[21]。
基于高通量测序平台的转录组测序技术能够全面获得物种特定组织或器官的转录本信息, 从而进行基因表达水平研究、新转录本发现研究、转录本结构变异研究等。Xu等[22]使用RNA-Seq对2个不同含油量的品种在3个发育阶段的油菜角果转录组进行分析, 鉴定了与油脂代谢相关的14个候选基因; Shah等[23]在不同发育阶段和温度条件下对半冬性双单倍体宁油7号进行了RNA测序(RzNA-seq), 鉴定出36个与开花时间、种子产量或两者相关的基因; Zhao等[24]利用2个高低含油量油菜品种不同发育时期种子转录组数据, 得到了18个与含油量相关的候选基因。
全基因组关联研究(Genome-Wide Association Studies, GWAS)被用作检测QTL/基因的替代方法, 并提供了一种高分辨率的图谱, 以识别目标性状的特定单核苷酸多态(Single Nucleotide Polymorphism, SNP), 从序列数据中生成的廉价、高密度的SNP标记目前可用来评估甘蓝型油菜的全基因组分析。He等[25]利用60k芸苔属Illumina Infinium SNP对327种质材料进行了分支数目的GWAS研究, 结果表明, 4个SNP位点与分枝数显著相关; Gajardo等[26]利用4025个SNP对89份油菜材料的硫苷和纤维素含量进行GWAS分析, 鉴定出17个SNP与种子硫苷含量相关, 5个SNP与纤维素含量显著相关; Liu等[27]利用Brassica 60K SNP对521份甘蓝型油菜种子含油量进行全基因组关联研究, 鉴定出50个与种子含油量显著相关的位点, 这些位点可以解释约80%的表型变异, 其中29个位点未被报道; Xiao等[28]对588份油菜自交系进行高通量基因组重测序获得的385,692个SNP, 并对含油量进行全基因组关联分析,共鉴定出17个与含油量显著相关的位点; Qu等[29]利用11,368个SNP标记, 对520份材料进行了油脂组成的全基因组关联研究, 最终预测24个参与油脂生物合成的候选基因; Zhao等[30]利用2,340,881个SNP对204份油菜材料进行GWAS分析, 鉴定出16个与油菜油含量显著相关的位点。因此, 全基因组关联分析在挖掘数量性状基因和检测功能基因有利变异位点具有独特的优势。
本研究以长沙地区4个甘蓝型油菜品种为研究对象, 利用转录组分析种子发育过程中基因的动态表达, 筛选出影响含油量的候选基因集。结合区域关联分析鉴定A10、A05基因结构变异影响了含油量积累。共表达网络分析表明,A10、A05、LEC1、HMGB3与HTA11形成分子网络, 共同对含油量的积累进行调控。这些结果能用于开发功能标记来选育高含油量品种。
1 材料与方法
1.1 试验材料
转录组分析选用甘蓝型油菜品种(系)为试验材料, 分别为CS115、CS136、XY015和XY777。4个试验材料在湖南长沙湖南农业大学耘园基地进行秋播。在油菜现蕾时, 每个材料选取5株形态、长势一致的植株, 标记出主花序并套袋自交, 在标记后第25、35及45天从每个标记单株取5个角果, 混合后将角果皮与种子在冰浴条件下尽快分离, 分离后的种子置于-80℃的超低温冰箱中保存, 以备提取RNA进行转录组测序。
区域关联分析选用50份中国半冬油菜材料, 来自于中国西南大学重庆油菜工程中心, 这些材料代表亚洲油菜的多样性。这些材料种植在德国大田和温室, 调查并整理含油量表型。2013年(Field_2013)和2014年(Field_2014)分别在Gross Gerau (49.94°N, 8.50°E)和Rauischholzhausen (50.76°N, 8.88°E)进行了无重复完全随机设计的大田试验。每株系都种植3行, 每行25株, 每行株间5 cm, 行间25 cm。采集各小区3株自花授粉种子, 测定种子含油量。2012年, 在德国尤斯图斯-李比希大学(Justus Liebig University Giessen, GH_2012)温室进行种植, 每个株系种植5株材料, 3株自交系种子用于测定含油量。用近红外光谱法测定了50份甘蓝型油菜种子含油量。
1.2 数据分析
1.2.1 转录组数据分析 CS115、CS136、XY015和XY777作为转录组分析的样品, 取发育25 d、35 d、45 d的油菜角果, 摘下后立即在液氮中冷冻并储存在-80℃直至RNA提取。采用天根生化科技有限公司的植物总RNA提取试剂盒DP432提取油菜种子的RNA。随后分别用NanoPhotometer分光光度计(IMPLEN, 美国)和Qubit3.0 Flurometer (Life Technologies, 美国)检测RNA纯度和浓度。然后使用Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, 美国)评估RNA的完整性。样品总RNA质量合格后, 对质量良好、完整性合格的RNA进行进一步处理, 清洗含poly-A mRNA片段, 合成双链cDNA, 通过聚合酶链反应(Polymerase Chain Reaction, PCR)进行扩增。cDNA测序文库有效浓度大于10 nmol L–1的样本被认为可以进行测序。测序采用Illumina HiSeq X测序平台, 测序策略为PE150 (对端2× 150 bp模式), 对原始数据采取各种质量控制措施, 获得高质量数据。所有随后的分析都是基于高质量数据。使用HISAT2将高质量reads与甘蓝型油菜参考基因组进行比对。使用HTSeq v0.6.1计算每个基因的reads数。然后, 根据基因的长度和reads计数, 计算每个基因的转录本每千碱基每百万reads的片段。
1.2.2 基因筛选和表达分析、GO功能富集分析
利用R软件中DEseq2程序包进行基因表达分析。基因筛选标准为: |log2(Fold Change)| > 1和< 0.05。将筛选的基因比对到拟南芥基因组数据库, 筛选与含油量相关基因, 用R软件heatmap包将候选基因集表达量进行可视化处理。采用TBtools软件进行基因的GO富集分析, 对每个有代表性的GOterm计算值,值小于0.05时GOterm被认为是显著富集的。GO功能显著性富集分析分别包括基因的分子功能(molecular function)、细胞组分(cellular component)、生物过程(biological process)。
1.2.3 重测序分析 对来自西南大学的50份多世代(自花授粉至少5代)半冬性甘蓝型油菜种质资源进行测序。采用十六烷基三甲基溴化铵(CTAB)的方法[31]从幼叶中提取基因组DNA。使用Illumina HiSeq 4000仪器(Illumina Co., Inc., 美国)构建DNA文库。生物标记技术公司(中国北京)完成文库的准备和测序。利用Burrows-Wheeler Aligner的MEM算法[32]将50个中国油菜自选系筛选后的reads定位到油菜参考基因组(https://www.genoscope.cns.fr/brassicanapus/)。用GATK对每个样品进行SNP调用(https://www.broadinstitute.org/gatk/guide/best-practices. php), 使用Beagle软件包[33]去除缺失率≥0.6的SNP。最后选用532,005个等位基因频率(MAF) ≥0.05、杂合率≥0.25的优质SNP标记, 用于油菜种子含油量的区域关联分析。
1.2.4 含油量的区域关联分析 50份中国半冬性甘蓝型油菜的全基因组重测序在Illumina HiSeq 4000平台完成, 测序共产生了28.6亿对reads, 每个材料的平均测序深度大约为5倍, 基因组覆盖率达到85%。50份中国半冬性甘蓝型油菜, 共检测320万个SNP位点。通过严格的过滤后, 533,245个高质量SNP标记被用种子含油量关联分析。利用TASSEL 5.0软件[34]和Q + K (Population Structure and Kinship)混合线性模型型(Mixed Linear Model, MLM)[35]对标记-性状关联进行检测。R软件包QQman[36]被用于绘制曼哈顿图和Quantile-Quantile散点图。FDR (False Discovery Rate)值的计算是利用R语言中的-value软件包(http://www.Bioconductor.org/packages/ release/bioc/qvalue.html), 最终确定显著水平的值(≤1×10–4)。
1.2.5 共表达网络分析 12份(4份甘蓝型油菜3个不同时期)中国半冬性甘蓝型油菜种质的转录组中获得基因表达数据。使用软件R中的R包“WGCNA”[37]构建共表达网络, 将权重参数的截止值设为0.2。输出文件后在Cytoscape 3.6版软件[38]中进行基因模块和关键基因网络的可视化。
2 结果与分析
2.1 油菜角果含油量表型统计和分析
对种植在长沙的4个油菜品种进行含油量表型统计和分析发现, CS115、CS136、XY015和XY777成熟种子含油量平均值±标准差分别为48.84±1.21 (%)、49.50±1.30 (%)、40.39±1.28 (%)、33.32±1.67 (%), 变异系数分别为2.32%、2.62%、3.16%、5.02%, 4个品种含油量具有差异性(表1)。
2.2 差异表达基因的统计
对所有基因进行表达量分析发现, 授粉后25~35 d, CS115、CS136、XY015和XY777分别有5513、7191、5537、5213个基因上调表达, 6166、8275、5107、5319个基因下调表达; 授粉后35~45 d, CS115、CS136、XY015和XY777分别有3900、4633、3655、3475个基因上调表达, 4347、5871、4145、4186个基因下调表达(图1-a)。统计4个品种授粉后25~45 d持续上调或下调的表达基因, 得到383个持续上调或下调表达基因, 其中持续上调表达基因292个, 持续下调表达基因91个(图1-b, c)。
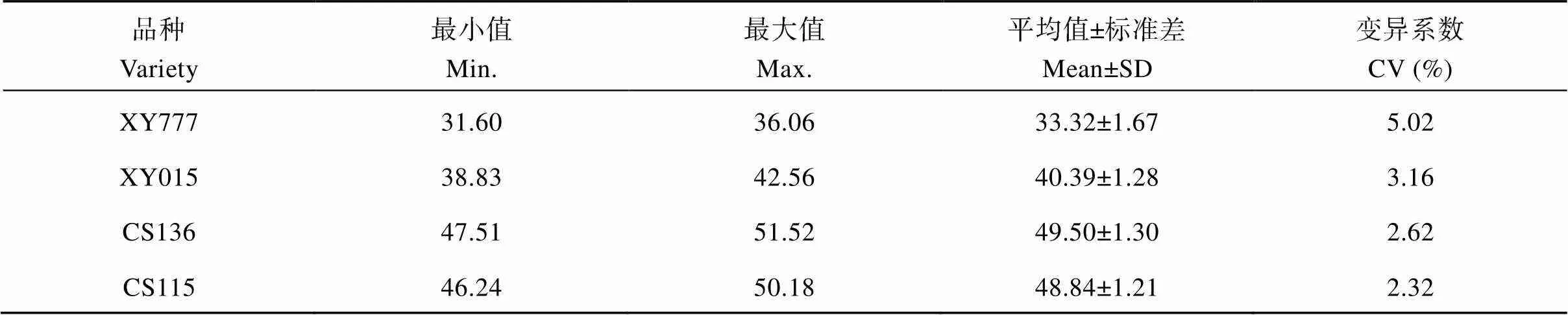
表1 长沙地区4个油菜品种成熟种子含油量统计表

图1 持续上调或下调表达基因筛选统计
(a) 长沙地区4个油菜品种, 25~35 d、35~45 d持续上调或下调基因数量统计图。(b) 4个品种持续上调基因维恩图。(c) 4个品种持续下调基因维恩图。
(a) the number of continuously up-regulated or down-regulated genes of four rapeseed varieties in Changsha area during 25–35 days and 35–45 days periods. (b) Venn diagrams of four varieties consistently up-regulated genes. (c) Venn diagrams of consistently down-regulated genes in four varieties.
2.3 基因通路富集分析
将筛选出的383个基因注释到GO数据库中, 进行通路富集分析(图2)发现, 大量基因在生化过程中参与油脂合成(GO:0033993), 部分差异基因与细胞组分中油脂小滴有关(GO:0005811), 说明初步筛选的基因功能与油脂合成具有关联。
2.4 基因表达分析
将383个基因在油菜Darmor-bzh参考基因组v.4.2中进行比对, 得到其基因序列后在拟南芥基因组数据库中确认基因功能, 得到43个与含油量相关基因。其中有33个基因上调表达, 动态分析显示授粉后25~45 d表达量持续升高, 尤其授粉后25~35 d表达量迅速升高, 包括-A09、-A02、C02、A04和A10,A05表达量授粉后35~ 45 d迅速升高; 10个基因下调表达, 动态分析显示在授粉后25~45 d表达量呈现下降趋势, 尤其授粉后35~45 d表达量迅速下降, 包括-A07-C07等含油量积累的主要转录调控因子(图3)。
圆圈大小表示基因数量, 热图表示–log10(-value)的值。
The size of the circle represents the number of genes, and the heat map represents the value of –log10(-value).
2.5 区域关联分析和单倍型分析
利用区域关联分析进一步解析这43个差异表达基因。含油量的区域关联分析采用Q+K模型, 基于-log10()≥4的显著截点, 检测到2个显著的单倍型A10 (1,969,139~1,969,452 bp,2=0.9993), A05 (4,414,567~ 4,416,061 bp,2=0.9985)与含油量显著相关, 进一步分析鉴定到2个候选基因(,-A10)、(,-A05)。3个SNP位于-A10启动子区(A10: 1969139; A10: 1969444; A10: 1969452,= 1.35×10–5) (图4-b)。在-A10基因区检测到2个单倍型(Hap)等位基因(图4-c), 对比分析这2个单倍型等位基因所对应的含油量表型发现,-A10_Hap1对应材料的含油量显著高于-A10_Hap2对应材料的含油量(检验-双样本等方差假设; 图4-d)。
在A05染色体的4,413,939~4,416,132 bp, 发现基因-A05 ()与种子含油量显著相关(图5-a)。9个SNP位于-A05基因区内, 1个SNP (A05: 4414567;=1.42×10–4)在基因外显子区, 与种子含油量显著相关(图5-b)。在- A05中鉴定出3个单倍型等位基因(图5-c), Hap1对应材料的含油量显著高于Hap3对应材料的含油量(检验-双样本等方差假设; 图5-d)。

图3 热图显示与含油量相关差异表达基因的表达量(DEGs)
RNA-seq数据的表达值进行了log10(fpkm+1)转换, 并显示为填充块, 从蓝色到黄色, 再到红色。
The expression values for RNA-seq data were log10(fpkm+1) transformed and displayed as filled blocks, from blue to yellow to red.

图4 50个重测序材料中单体型(1,944,128~1,994,025 bp)区域含油量关联分析
(a) 单体型(1,944,128~1,994,025 bp;2=0.99)区域的含油量关联分析。蓝色实线表示全基因组显著性的阈值值为1.0×10–4。(b)和(c) 3个SNP (Chr.A10: 1,969,139; Chr.A10: 1,969,444; Chr.A10: 1,969,452,=1.35×10–5)与含油量显著相关, 并定位在-A10基因启动子区域。热图显示这些SNPs存在强的连锁不平衡。在-A10单倍型区检测到2个单倍型等位基因。(d) 比较分析2个单体型等位基因对应材料的含油量。单体型等位基因在群体中的频率大于0.01将被用于此分析。箱型图显示-A10_Hap1等位基因对应的材料含油量显著高于-A10_Hap2对应的材料含油量。*、**、***分别表示在0.05、0.01和0.001概率水平差异显著。
(a) Haplotype (1,944,128–1,994,025 bp;2=0.99) oil content correlation analysis. The solid blue line indicates a threshold-value of 1.0×10–4for genome-wide significance. (b)–(c) three SNPs (Chr. A10: 1,969,139; Chr. A10: 1,969,444; Chr. A10: 1,969,452,= 1.35×10–5) was significantly correlated with oil content and was localized in the promoter region of-A10 gene. Heat maps show a strong linkage imbalance in these SNPs. Two haplotype alleles were detected in the-A10 haplotype region. (d) Comparative analysis of the oil content of the materials corresponding to the two haplotype alleles. Haplotype alleles with frequencies greater than 0.01 in the population will be used for this analysis. The box pattern shows that the material corresponding to the-A10_Hap1 allele has a higher oil content than that of-A10_Hap2. *, **, and *** mean significant difference at the 0.05, 0.01, and 0.001 probability levels, respectively.
2.6 共表达网络分析
为进一步分析A10和-A05基因功能联系, 本研究利用12份甘蓝型油菜种子转录组数据构建了共表达网络。该分析产生了4个基因模块, 每个模块在输出中用不同的颜色表示(图6-a)。模块包含候选基因已被WGCNA识别,-A10和-A05都位于与含油量相关的绿松石模块中(图6-c)。在功能注释的基础上, 将共表达网络基因进行功能分类, 共有113个基因位于亚网络中, 与油脂生物合成过程、油脂转运、油脂氧化、光合作用、碳水化合物代谢过程、转录因子相关基因分别有30、15、11、12、18、27个(图6-d)。与直接相连, 另外与通过3个转录因子LEC1、HMGB3、HTA11间接相连,- A10、-A05和众多影响油脂代谢基因形成了种子油脂积累潜在调控的分子网络。

图5 50个重测序材料中单体型(4,389,567~4,439,432 bp)区域的含油量关联分析
(a) 单体型(4,389,567~4,439,432 bp;2=0.99)区域的含油量关联分析。蓝色实线表示全基因组显著性的阈值值为1.0×10–4。(b)和(c) 9个SNP (A05: 4414567;=1.42×10–4)与含油量显著相关, 并定位在-A05基因区域。热图显示这些SNPs存在强的连锁不平衡。在-A05单倍型区检测到3个单倍型等位基因。(d) 比较分析3个单体型等位基因对应材料的含油量。单体型等位基因在群体中的频率大于0.01将被用于此分析。箱型图显示-A05_Hap1等位基因对应的材料有较高的含油量。*、**、***分别表示在0.05、0.01和0.001水平差异显著。
(a) Haplotype (4,389,567–4,439,432 bp;2=0.99) oil content correlation analysis. The solid blue line indicates a threshold-value of 1.0×10–4for genome-wide significance. (b)–(c) nine SNPs (A05: 4,414,567;= 1.42×10–4) was significantly correlated with oil content and was localized in the-A05 gene region. Heat maps show a strong linkage imbalance in these SNPs. Three haplotype alleles were detected in the haplotype region of-A05. (d) Comparative analysis of the oil content of the materials corresponding to the three haplotype alleles. Haplotype alleles with frequencies greater than 0.01 in the population will be used for this analysis. The box pattern shows that the material corresponding to the-A05_Hap1 allele has a higher oil content. *, **, and *** mean significant difference at the 0.05, 0.01, and 0.001 probability levels, respectively.
3 讨论
高含油量油菜的选育一直是油菜育种的主要目标之一, 然而, 油菜含油量是一个复杂的数量性状, 受多种遗传和环境因素的调控[39]。现有的高通量基因分型技术促进了GWAS对复杂性状的解析。最近有研究者对505、370和204份具有高密度SNP的不同油菜品系进行了GWAS和转录组数据分析, 分别得到27、7和18个SNP与种子含油量显著相关, 筛选出重要候选基因[40-42]。本研究利用转录组数据初步筛选到383个差异表达基因, 比对拟南芥基因功能数据库, 其中有43个与含油量相关差异表达基因, 包括-A09、-A02、- C02、-A04、-A10、-A07、-C07和-A05等重要的油脂合成代谢相关基因。紧接着对这43个基因进行区域关联分析和单倍型分析发现,-A10和-A05基因与含油量显著相关, 其中检测到2个优良的单体型等位基因(-A10_Hap1和-A05_ Hap1)。在拟南芥中,是5个编码油体蛋白的基因之一, 拟南芥突变会导致成熟种子含油量低于对照组[43]。ABI5能够调控编码TAG (三酰基甘油)生物合成的(二酰基甘油酰基转移酶)表达, 进而影响拟南芥种子含油量[44]; ABI5还能激活WRI1-1 (编码激活油脂生物合成基因表达)的转录, 拟南芥WRI1-1超表达株系种子含油量得到提高[45]。此外, 在共表达基因网络中,与通过3个转录因子LEC1、HMGB3、HTA11间接相连。有研究表明ABI3调节的表达[46], 并且ABI3与ABI5联系密切, 能够增强彼此表达[47], 所以ABI5可能参与的表达调控, 结合共表达网络分析和前人研究, 本研究推测参与等基因组成的潜在分子网络调控含油量的积累。这些结果有利于我们开发单体型功能标记, 进一步提高油菜含油量。

图6 共表达网络分析
(a) 模块系统树图。(b) 模块与含油量相关性。(c) 模块中基因数目对比。(d) 基因网络图。八边形红色节点代表候选基因, 根据功能标注, 共表达网络分为油脂生物合成过程(红色节点)、油脂转运(紫色节点)、油脂氧化(橙色节点)、光合作用(绿色节点)和碳水化合物代谢过程(灰色节点)。
(a): dendrogram of module system. (b): correlation between modules and oil content. (c): comparison of gene numbers in modules. (d): gene network diagram. Octagonal red nodes represent candidate genes, and according to functional labeling, co-expression networks are divided into: lipid/fatty acid biosynthesis (red nodes), lipid transport (purple nodes), lipid/fatty acid oxidation (orange nodes), photosynthesis (green nodes), and carbohydrate metabolism (gray nodes).
4 结论
利用转录组数据筛选到43个与含油量相关的持续上调或下调表达基因, 包括、、和等。结合区域关联分析, 检测到43个基因中的和与含油量存在显著关系, 并鉴定到-A10_Hap1和-A05_Hap1单倍型等位基因对应材料的含油量较高。另外, 结合共表达网络分析揭示与通过3个转录因子LEC1、HMGB3、HTA11间接相连形成潜在的网络影响含油量积累。本研究结果可为油菜含油量改良奠定基础, 为进一步提高含油量提供理论依据。
[1] Liu S, Fan C, Li J, Cai G, Yang Q, Wu J, Yi X, Zhang C, Zhou Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of., 2016, 129: 3–15.
[2] Hua W, Liu J, Wang H. Molecular regulation and genetic improvement of seed oil content inL., 2016, 3: 186–194.
[3] 王汉中. 未来15年中国油菜遗传改良策略. 中国油料作物学报, 2004, 26: 98–101. Wang H Z. Strategy for rapeseed genetic improvement in China in the coming fifteen years., 2004, 26: 98–101 (in Chinese with English abstract).
[4] Xin F W, Gui H L, Qing Y, Wei H, Jing L, Wang H Z. Genetic analysis on oil content in rapeseed (L.)., 2010, 173: 17–24.
[5] Hua W, Li R J, Zhan G M, Liu J, Li J, Wang X F, Liu G H, Wang H Z. Maternal control of seed oil content in: the role of silique wall photosynthesis., 2012, 69: 32–44.
[6] Yan L G, Ping S, Nan W, Jing W, Bin Y, Chao Z M, Jin X T, Ji T Z, Ting D F, Jin X S. Genetic effects and genotype × environment interactions govern seed oil content inL., 2017, 18: 1.
[7] Jing L, Wei H, Hong L Y, Gao M Z, Rong J L, Lin B D, Xin F W, Gui H L, Wang H Z. Thegene (-like gene from) enhances seed oil production through regulating cell number and plant photosynthesis., 2012, 63: 3727–3740.
[8] Chao H, Wang H, Wang X, Guo L, Gu J, Zhao W, Li B, Chen D, Raboanatahiry N, Li M. Genetic dissection of seed oil and protein content and identification of networks associated with oil content in., 2017, 7: 46295.
[9] Shi J, Lang C, Wang F, Wu X, Liu R, Zheng T, Zhang D, Chen J, Wu G. Depressed expression ofandgenes modifies fatty acid profiles and storage compounds accumulation inseeds., 2017, 263: 177–182.
[10] Frentzen M. Acyltransferases from basic science to modified seed oils., 2010, 100: 161–166.
[11] Hills M J. Control of storage-product synthesis in seeds., 2004, 7: 302–308.
[12] Bates P D, Johnson S R, Cao X, Li J, Nam J W, Jaworski J G, Ohlrogge J B, Browse J. Fatty acid synthesis is inhibited by inefficient utilization of unusual fatty acids for glycerolipid assembly., 2014, 111: 4–9.
[13] Turnham E, Northcote D H. Changes in the activity of acetyl-CoA carboxylase during rape-seed formation., 1983, 212: 3–9.
[14] Weselake R J, Shah S, Tang M, Quant P A, Snyder C L, Furukawa-Stoffer T L, Zhu W, Taylor D C, Zou J, Kumar A, Hall L, Laroche A, Rakow G, Raney P, Moloney M M, Harwood J L. Metabolic control analysis is helpful for informed genetic manipulation of oilseed rape () to increase seed oil content., 2008, 59: 3–9.
[15] Lock Y Y, Snyder C L, Zhu W, Siloto R M, Weselake R J, Shah S. Antisense suppression of type 1 diacylglycerol acyltransferase adversely affects plant development in., 2009, 137: 61–71.
[16] Kagaya Y, Toyoshima R, Okuda R, Usui H, Yamamoto A, Hattori T.controls seed storage protein genes through its regulation ofand., 2005, 46: 399–406.
[17] Wang H, Guo J, Lambert K N, Lin Y. Developmental control ofseed oil biosynthesis., 2007, 226: 73–83.
[18] Wu X L, Liu Z H, Hu Z H, Huang R Z.coordinates fatty acid biosynthesis and photosynthesis pathways during oil accumulation in rapeseed., 2014, 56: 82–93.
[19] Elahi N, Duncan R W, Stasolla C. Decreased seed oil production inmutant plants., 2015, 96: 22–30.
[20] Elahi N, Duncan R W, Stasolla C. Modification of oil and glucosinolate content in canola seeds with altered expression of., 2016, 100: 52–63.
[21] Wang Z, Qiao Y, Zhang J, Shi W, Zhang J. Genome wide identification of microRNAs involved in fatty acid and lipid metabolism ofby small RNA and degradome sequencing., 2017, 619: 61–70.
[22] Xu H M, Kong X D, Chen F, Huang J X, Lou X Y, Zhao J Y. Transcriptome analysis ofpod using RNA-Seq and identification of lipid-related candidate genes., 2015, 16: 858.
[23] Shah S, Weinholdt C, Jedrusik N, Molina C, Zou J, Große I, Schiessl S, Jung C, Emrani N. Whole-transcriptome analysis reveals genetic factors underlying flowering time regulation in rapeseed (L.)., 2018, 41: 1935–1947.
[24] Zhao C, Xie M, Liang L, Yang L, Han H, Qin X, Zhao J, Hou Y, Dai W, Du C, Xiang Y, Liu S, Huang X. Genome-wide association analysis combined with quantitative trait loci mapping and dynamic transcriptome unveil the genetic control of seed oil content inL., 2022, 13: 929197.
[25] He Y, Wu D, Wei D, Fu Y, Cui Y, Dong H, Tan C, Qian W. GWAS, QTL mapping and gene expression analyses inreveal genetic control of branching morphogenesis., 2017, 7: 15971.
[26] Gajardo H, Wittkop B, Soto-Cerda B, Higgins E, Parkin I, Snowdon R, Federico M, Iniguez-Luy F. Association mapping of seed quality traits inL. using GWAS and candidate QTL approaches., 2015, 35: 143.
[27] Liu S, Fan C, Li J, Cai G, Yang Q, Wu J, Yi X, Zhang C, Zhou Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of., 2016, 129: 3–15.
[28] Xiao Z, Zhang C, Tang F, Yang B, Zhang L, Liu J, Huo Q, Wang S, Li S, Wei L, Du H, Qu C, Lu K, Li J, Li N. Identification of candidate genes controlling oil content by combination of genome-wide association and transcriptome analysis in the oilseed crop., 2019, 12: 216.
[29] Cun M Q, Le D J, Fu Y F, Hui Y Z, Kun L, Li J W, Xin F X, Ying L, Shi M L, Rui W, Jia N L. Genome-wide association mapping and identification of candidate genes for fatty acid composition inL. using SNP markers., 2017, 18: 232.
[30] Zhao C, Xie M, Liang L, Yang L, Han H, Qin X, Zhao J, Hou Y, Dai W, Du C, Xiang Y, Liu S, Huang X. Genome-wide association analysis combined with quantitative trait loci mapping and dynamic transcriptome unveil the genetic control of seed oil content inL., 2022, 13: 929197.
[31] Uzunova M, Ecke W, Weissleder K, Röbbelen G. Mapping the genome of rapeseed (L.): I. Construction of an RFLP linkage map and localization of QTLs for seed glucosinolate content., 1995, 90: 194–204.
[32] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform., 2009, 25: 1754–1760.
[33] Browning B L, Browning S R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals., 2009, 84: 10–23.
[34] Bradbury P J, Zhang Z, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23: 3–5.
[35] Lu K, Peng L, Zhang C, Lu J, Yang B, Xiao Z, Liang Y, Xu X, Qu C, Zhang K, Liu L, Zhu Q, Fu M, Yuan X, Li J. Genome-wide association and transcriptome analyses reveal candidate genes underlying yield-determining traits in., 2017, 8: 206.
[36] Turner S D. QQman: an R package for visualizing GWAS results using QQ and manhattan plots., 2014, 005165.
[37] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559.
[38] Shannon P, Markiel A, Ozier O, Baliga N S, Wang J T, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks., 2003, 13: 498–504.
[39] Si P, Mailer R J, Galwey N, Turner D W. Influence of genotype and environment on oil and protein concentrations of canola (L.) grown across southern Australia., 2003, 54: 397–407.
[40] Tang S, Zhao H, Lu S, Yu L, Zhang G, Zhang Y, Yang Q Y, Zhou Y, Wang X, Ma W, Xie W, Guo L. Genome- and transcriptome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in., 2021, 14: 470–487.
[41] Tang M Q, Zhang Y Y, Liu Y Y, Tong C B, Cheng X H, Zhu W, Li Z Y, Huang J Y, Liu S Y. Mapping loci controlling fatty acid profiles and oil and protein content by genome-wide association study in., 2019, 7: 217–226.
[42] Zhao C, Xie M, Liang L, Yang L, Han H, Qin X, Zhao J, Hou Y, Dai W, Du C, Xiang Y, Liu S, Huang X. Genome-wide association analysis combined with quantitative trait loci mapping and dynamic transcriptome unveil the genetic control of seed oil content inL., 2022, 13: 929197.
[43] López-Ribera I, La Paz J L, Repiso C, García N, Miquel M, Hernández M L, Martínez-Rivas J M, Vicient C M. The evolutionary conserved oil body associated proteinparticipates in the regulation of oil body size., 2014, 164: 37–49.
[44] Kong Y, Chen S, Yang Y, An C. ABA-insensitive (ABI)andsynergistically regulateexpression inseedlings under stress., 2013, 587: 76–82.
[45] Yeap W, Lee F L, Shan D, Musa H, Appleton D R, Kulaveerasingam H.,,andincrease oil biosynthesis in coordination with hormonal signaling during fruit development in oil palm., 2017, 91: 97–113.
[46] Crowe A J, Abenes M, Plant A, Moloney M M. The seed-specific transactivator,, induces oleosin gene expression., 2000, 151: 171–181.
[47] Nakamura S, Lynch T J, Finkelstein R R. Physical interactions between ABA response loci of., 2001, 26: 27–35.
A combination of genome-wide association and transcriptome analysis reveal candidate genes affecting seed oil accumulation in
CAO Song, YAO Min, REN Rui, JIA Yuan, XIANG Xing-Ru, LI Wen, HE Xin, LIU Zhong-Song, GUAN Chun-Yun, QIAN Lun-Wen*, and XIONG Xing-Hua*
College of Agronomy, Hunan Agricultural University, Changsha 410128, Hunan, China
Rapeseed (L.) is the main source of edible vegetable oil in China, and increasing seed oil content is the most effective way to increase the supply of rapeseed oil. In this study, 43 genes related to lipid synthesis were selected by analyzing the seed transcriptome data of 4 rapeseed inbred lines 25, 35, and 45 days after pollination. Among them, 33 genes were continuously up-expressed and 10 genes were continuously down-expressed from 25 to 45 days of seed development. The main genes included,,, and. At the same time, combined with the resequencing data of 50 semi-winter, 3 SNPs and 9 SNPs significantly related to oil content were detected to-A10 and-A05, respectively, and the oil content of-A10_Hap1 was significantly higher than Hap2. The oil content of-A05_Hap1 was significantly higher than Hap3. In addition, WGCNA was used to construct gene networks, and we found thatandwere indirectly connected through three transcription factors LEC1, HMGB3, and HTA11, which together formed a molecular network involved in the potential regulation of seed oil accumulation. The results of this study provide valuable insights for the development of haplotype functional markers, aiming to further enhance oil content in.
; oil content; transcriptome analysis; coexpression analysis; regional association analysis
10.3724/SP.J.1006.2024.34152
本研究由湖南省杰出青年科学基金项目(2022JJ10027), 湖南省教育厅科学研究重点项目(21A0135)和国家科技重大专项项目(2022ZD04009)资助。
This study was supported by the Science Foundation for Distinguished Youth Scholars of Hunan Province, China (2022JJ10027), the Research Foundation of Education Bureau of Hunan Province, China (21A0135), and the National Key Science and Technology Project (2022ZD04009).
熊兴华, E-mail: xiongene@hunau.edu.cn; 钱论文, E-mail: qianlunwen@163.com
E-mail: c18890046232@163.com
2023-09-08;
2024-01-12;
2024-01-25.
URL: https://link.cnki.net/urlid/11.1809.S.20240125.0905.002
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
