CpOGAD298N 与核心链霉亲和素Stv13融合表达用于检测蛋白质O-GlcNAc 修饰
2024-04-23黎先感张连文
黎先感, 杨 明, 鲁 莹, 李 玲, 马 骞, 李 静, 张连文*
(1)南开大学药学院, 天津分子药物研究重点实验室 300350, 天津;2)首都师范大学生命科学学院,DNA损伤应答北京市重点实验室 100048, 北京)
O-GlcNAc修饰是一种动态的翻译后修饰,发生在细胞内多种蛋白质的丝氨酸或苏氨酸残基上[1]。O-GlcNAc修饰由2种酶催化完成:OGT(O-GlcNAc transferase)将糖供体UDP-GlcNAc中的GlcNAc基团转移到蛋白质的丝氨酸或苏氨酸残基上[2];OGA(O-GlcNAcase)催化O-糖苷键水解,去除 GlcNAc 基团[3]。O-GlcNA修饰现已被证明参与许多生理过程,包括转录、翻译、蛋白质酶体降解和信号转导[4, 5]。它还通过稳定新生多肽链参与蛋白质折叠[6]。同时,O-GlcNAc蛋白的失调与许多慢性疾病有关,包括癌症、糖尿病、心血管并发症和阿尔茨海默病[7, 8]。
O-GlcNAc修饰的检测具有重要意义。在过去的数十年间,已经开发了多种检测方法,涵盖β-消除-米氏加成法[9, 10]、化学/酶法[11-13]、代谢标记法[14]、O-GlcNAc特异性抗体(如CTD110.6、RL2)和凝集素(s-WGA)[15-18],但它们均有各自的缺陷且在进一步区分GlcNAc和N-乙酰半乳糖胺(N-acetylgalactosamine,GalNAc)上仍然面临挑战[19]。
CpOGA来自产气荚膜羧菌,是真核生物OGA的细菌同源物,同样可以催化O-GlcNAc 糖蛋白去糖基化。尽管目前尚不清楚CpOGA是否是一种生理上的OGA,但它在体外对人类细胞系裂解物中的O-GlcNAc蛋白显示出显著的O-GlcNAcase活性[20]。研究发现,其突变体 CpOGAD298N丧失催化活性,保留了结合O-GlcNAc蛋白的能力,在O-GlcNAc蛋白的富集和检测方面具有明显优势[21, 22]。Song等人通过将谷胱甘肽S-转移酶(GST)标签与 CpOGAD298N融合表达,发现GST- CpOGAD298N可以在短时间(0.5 h)内结合膜上的O-GlcNAc蛋白,然后通过辣根过氧化物酶(horseradish peroxidase,HRP)标记的抗GST的二抗进行曝光显影[23]。而且CpOGAD298N相较O-GlcNAc抗体或凝集素具有更全面的O-GlcNAc蛋白结合图谱,并能区分GlcNAc和GalNAc的2种单糖修饰。但是,此方法与常规Western 印迹方法均存在耗时过长的问题(~36 h)。
生物素-亲和素系统是一种应用多年的生物反应放大系统[24, 25]。从链霉菌(streptomyces avidinii)培养物中提取的链霉亲和素(streptavidin,SA),与亲和素有相似的生物学特性,可以高度特异性地与D-生物素结合,解离常数处于10-14mol/L数量级[26-28]。相对于亲和素,SA有接近中性的等电点且不含任何糖基,检测灵敏度更好[29]。核心链霉亲和素是SA的衍生物,这种链霉亲和素仍可结合生物素分子,并且比全长链霉亲和素稳定性更好,溶解度更高,凝聚更难[30]。后来有学者通过基因重组,表达获得了118个氨基酸残基的Stv13核心链霉亲和素,Stv13具有与天然链霉亲和素相似的生物活性和稳定性,并且溶解性更高[31]。
本文构建了pET28a-CpOGAD298N-Stv13重组表达质粒,表达纯化了CpOGAD298N-Stv13融合蛋白质,并应用生物素-亲和素结合特点建立了一种O-GlcNAc蛋白快速检测方法。
1 材料与方法
1.1 材料
研究所用pET28a质粒、pMAL-c2x-sOGT质粒、FR_HNF1A质粒、HEK293T细胞为本实验室留存[32-34];E.coliDH5α/E.coliBL21(DE3)菌株、ECL化学发光超敏显色试剂盒、高保真PCR预混液购于上海翊圣生物科技有限公司;生物素购于MCE公司;生物素修饰的辣根过氧化物酶(biotin-HRP)(A0308)购于上海碧云天生物技术有限公司;限制性内切酶购于赛默飞世尔科技公司;RL2、c-Myc、β-肌动蛋白(β-Actin)抗体购于Abcam;二抗(Goat-anti-Rabbit、Goat-anti-Mouse)购于天津三箭生物技术有限公司;T4连接酶购于NEB;引物(Table 1)由北京擎科生物合成。

Table 1 Primers used for mutations
1.2 CpOGAD298N-Stv13融合蛋白质表达质粒构建
1.2.1 化学合成带有编码CpOGA的DNA序列 N末端及C末端分别带有EcoRⅠ、HindⅢ酶切位点,通过限制性内切酶EcoRⅠ、Hind Ⅲ 双酶切将其克隆到原核表达载体pET28a上,构建重组表达载体pET28a-CpOGA。
1.2.2 pET28a-CpOGAD298N重组载体的构建 以pET28a-CpOGA序列为模板,利用引物 pET28a-CpOGA-D298N(F/R)进行PCR构建pET28a-CpOGAD298N重组表达载体。
1.2.3 化学合成带有编码(G4S)4-Stv13的DNA序列 N末端及C末端分别带有Hind Ⅲ、XhoⅠ 酶切位点,通过限制性内切酶Hind Ⅲ、XhoⅠ双酶切,将其克隆到pET28a-CpOGAD298N上,构建重组表达载体pET28a-CpOGAD298N-Stv13。
将pET28a-CpOGAD298N-Stv13重组表达载体转入E.coli DH5α 感受态细胞中,抗性平板筛选转化菌落,选取单克隆提取质粒,质粒通过测序和限制性内切酶EcoRⅠ、XhoⅠ 双酶切鉴定,质粒设计构建见Fig.1。
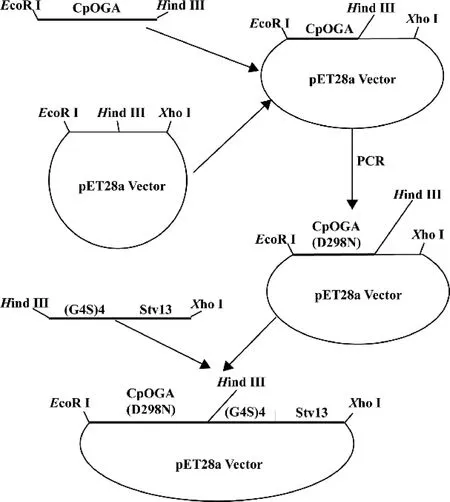
Fig.1 Schematic diagram of the plasmid construction The pET28a vector and chemically synthesized DNA sequences encoding CpOGA and (G4S) 4-Stv13 were used. The pET28a-CpOGAD298N-Stv13 plasmid was constructed by the cloning method of T4 DNA ligase ligation after corresponding restriction endonuclease digestion and site-directed mutagenesis PCR
1.3 蛋白质的诱导表达
将pET28a-CpOGA,pET28a- CpOGAD298N-Stv13,pMAL-c2x-sOGT(表达产物为MBP-sOGT)质粒分别转化进E.coliBL21(DE3)菌株中,涂板,37 ℃培养过夜后挑取单克隆菌落至加有相应抗生素的LB液体培养基中,37 ℃,220 r/min培养过夜;第2 d将菌液按1∶50比例扩大培养,37 ℃,220 r/min,测A600=0.7~0.9时,加入IPTG诱导表达24 h;收菌,保存在-80 ℃。
1.4 蛋白质的纯化
镍离子亲和层析纯化His标签蛋白质:使用15 mL 含20 mmol/L咪唑的TBS溶液(pH=7.4)重悬菌体,冰水浴中超声破碎16 min (开2 s,停4 s);4 ℃,12 000 r/mim,离心30 min后取上清;通过含20 mM咪唑的TBS溶液平衡镍离子亲和柱,取上清液通过Ni-NTA亲和柱结合,使用20 mmol/L咪唑溶液流过亲和柱除杂质,再用含250 mmol/L咪唑的TBS溶液洗脱目标蛋白质,最后通过30 kD截流分子量的超滤管(MilliporeSigma)超滤浓缩蛋白质。
淀粉树脂亲和层析纯化MBP标签蛋白质:使用15 mL 1×TBS溶液重悬菌体,冰水浴中超声破碎16 min(开2 sec,停4 sec);4 ℃,12 000 r/min,离心30 min后取上清;上清过直链淀粉-琼脂糖介质后用1×TBS溶液洗杂质,10 mmol/L麦芽糖溶液洗脱蛋白质,通过30 kD截流分子量的超滤管(MilliporeSigma)超滤浓缩蛋白质。
1.5 MBP-sOGT的去O-GlcNAc修饰
在PBS溶液中,将10 mg纯化后的MBP-sOGT蛋白质与2 mg CpOGA蛋白混合,室温反应1 h,获得去O-GlcNAc修饰的MBP-sOGT,即MBP-sOGT-DG,通过淀粉树脂亲和层析柱纯化MBP-sOGT-DG蛋白质。
1.6 蛋白质染色鉴定
所有蛋白质样本通过8 %的SDS-PAGE进行分离,恒压90 V,30 min后,恒压90 V,100 min,电泳结束后通过考马斯亮蓝染色法显示蛋白质条带。
1.7 细胞培养
人胚胎肾细胞 HEK293T使用含有10 %胎牛血清和1 %青链双抗的DMEM培养液,放置于37 ℃,5 % CO2的细胞培养箱中培养。待其达到80 % ~ 90 %的汇合度时,以1∶3的比例进行传代培养。向细胞中加入终浓度为5 μmol/L的TMG溶液,增加细胞糖基化,24 h后收取细胞进行裂解,BCA定量检测。
1.8 HNF1A蛋白质样品制备
在人胚胎肾细胞HEK293T中过表达FR_HNF1A质粒,48 h后收取细胞裂解,获得细胞裂解液;提前取200 μL 蛋白A与2 μL Myc-Tag抗体于PBS溶液中结合,4 ℃,过夜;第2 d对蛋白A进行洗涤,加入细胞裂解液进行IP,4 ℃,3 h后洗涤蛋白A,吸干上清,加入40 μL 1×上样缓冲液(loading buffer)煮沸,离心后上清即为IP得到的HNF1A样品。
1.9 O-GlcNAc修饰的检测
O-GlcNAc修饰的检测通过常规Western 印迹检测或改进型Far-WB实验完成。
Western 印迹:蛋白质样品制备后, 通过8 %SDS-PAGE胶进行分离,电泳结束后将SDS-PAGE胶上的蛋白质条带转移到PVDF膜上,5 % BSA室温封闭2 h,加入RL2(1∶1 000)作为一抗4 ℃孵育过夜,TBST缓冲液洗涤3次,加入二抗(1∶5 000)室温孵育2 h,TBST洗膜3次,使用化学发光试剂显影曝光。
Far-WB实验:经上述转膜后,用5 % BSA(含有20 μg/mL生物素)封闭2 h,TBST洗膜3×3 min;将1 mg CpOGAD298N- Stv13与5 μL生物素修饰的辣根过氧化物酶(biotin-HRP)混合,并用含5 % BSA的TBST稀释到4 mL,4 ℃孵育PVDF膜1 h;TBST洗膜5×8 min,使用化学发光试剂显影曝光;实验检测原理见Fig.2。

Fig.2 Schematic diagram of the far-WB detection principle The fusion protein CpOGAD298N-Stv13 captures the O-GlcNAc protein on the membrane by CpOGAD298N. Then the biotin-avidin strong interaction makes Stv13 to stably bind to biotin-HRP, and then chemiluminescence can be used
2 结果
2.1 pET28a-CpOGAD298N -Stv13重组质粒的鉴定
依据1.2方法构建pET28a-CpOGAD298N-Stv13重组质粒,质粒图谱见Fig.3A。构建后的重组质粒通过双酶切(EcoRⅠ、XhoⅠ)方法进行酶切鉴定,酶切预期分子量为2 196 bp和5 290 bp,大小均与预期相符,电泳结果见Fig.3B。
2.2 融合蛋白质CpOGAD298N -Stv13的纯化
将表达有 CpOGAD298N-Stv13融合蛋白质的BL21(DE3)菌体进行超声破碎,离心所得上清通过镍离子亲和层析柱纯化,离心后上清、沉淀、穿透及纯化后的蛋白质进行SDS-PAGE 分析。结果表明,CpOGAD298N-Stv13在E.coli BL21(DE3)菌株中可溶性表达, 蛋白质分子量约90 kD,与理论值(83 kD)一致(Fig.4A)。

Fig.4 Analysis of protein purification by SDS-PAGE (A) Purification of CpOGAD298N-Stv13; Lane 1, Induced cells; Lane 2, Centrifugal supernatant of induced cells after ultrasonication; Lane 3, Centrifugal precipitation of induced cells after ultrasonication; Lane 4, Flow-through solution of the Ni-NTA column after sampling; Lane 5, Ni-NTA column eluent after ultrafiltration concentration. (B) Purification of CpOGA; Lane 1,Induced cells;Lane 2, Centrifugal supernatant of induced cells after ultrasonication; Lane 3, Centrifugal precipitation of induced cells after ultrasonication; Lane 4, Ni-NTA column eluent after ultrafiltration concentration. (C) Purification of MBP-sOGT; Lane 1, Centrifugal supernatant of induced cells after ultrasonication; Lane 2, Centrifugal precipitation of induced cells after ultrasonication; Lane 3, Induced cells; Lane 4, Flow-through solution of the starch resin column after sampling; Lane 5, The starch resin column eluent after ultrafiltration concentration. (D) Purification of MBP-sOGT-DG; Lane 1, Centrifugal supernatant of induced cells after ultrasonication; Lane 2, Flow-through solution of the starch resin column after sampling; Lane 3, The starch resin column eluent after ultrafiltration concentration
2.3 底物蛋白质的纯化
本课题组前期研究结果表明,sOGT在E.coliBL21(DE3)表达时可发生自身O-GlcNAc修饰,且12位丝氨酸(Thr-12)是其主要修饰位点[32]。为了给sOGT蛋白,去O-GlcNAc修饰,本文表达纯化了具有O-GlcNAcase活性的CpOGA蛋白,分子量为75 kD(Fig.4B)。同时,为了方便去O-GlcNAc修饰的sOGT蛋白纯化,选择 pMAL-c2x-sOGT质粒表达MBP-sOGT融合蛋白质。将纯化后的MBP-sOGT分成2份,1份含有O-GlcNAc修饰直接保留,另1份经CpOGA去O-GlcNAc修饰后通过淀粉树脂亲和层析柱再次纯化,得到MBP-sOGT-DG蛋白。SDS-PAGE电泳结果表明:MBP-sOGT蛋白大部分以包涵体形式存在,可溶性表达较低,分子量为110 kD(Fig.4C,D)。
2.4 底物蛋白质O-GlcNAc修饰检测
应用Western 印迹方法对相同剂量的MBP-sOGT和MBP-sOGT-DG蛋白质进行O-GlcNAc修饰检测,结果显示,MBP-sOGT 具有糖基化修饰,MBP-sOGT-DG蛋白,未检测出糖基化修饰(Fig.5A),表明CpOGA成功去除了MBP-sOGT 的O-GlcNAc修饰,MBP-sOGT和MBP-sOGT-DG可以作为O-GlcNAc修饰检测的阳性和阴性对照。
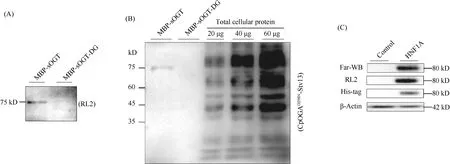
Fig.5 Detection of O-GlcNAc proteins at the cellular level (A) Routine WB assay using the anti-O-GlcNAc antibody RL2. (B) The Far-WB assay using CpOGAD298N -Stv13. The total cellular protein was extracted from HEK293T cells. (C)Routine WB assay using the anti-O-GlcNAc antibody RL2, c-Myc and β-Actin. The Far-WB assay using CpOGAD298N -Stv13. HNF1A was extracted from HEK293T cells
2.5 通过Far-WB方法检测蛋白质O-GlcNAc修饰
以MBP-sOGT、MBP-sOGT-DG为对照,使用CpOGAD298N-Stv13对HEK293T细胞总蛋白质O-GlcNAc修饰进行了检测。同时,为了研究本文条件下CpOGAD298N-Stv13检测蛋白质O-GlcNAc修饰与蛋白质剂量的线性关系,在电泳上样时加入不同剂量的HEK293T细胞裂解液上清蛋白质。Far-WB结果显示,CpOGAD298N-Stv13检测对照底物蛋白质O-GlcNAc修饰与RL2效果一致,证明了该Far-WB 方法的可靠性(Fig.5B)。同时,不同剂量细胞总蛋白质检测结果显示:随着剂量的升高所检测出条带灰度随之升高(Fig.5B),40 μg总蛋白质是比较适中的上样量,结果条带清晰,有较好的分辨率。
HNF1A(hepatocyte fuclear factor 1A)是一种广泛存在的转录因子,与肝的发育和维持密切相关。本课题组前期研究发现,HNF1A蛋白可被O-GlcNAc修饰,且该修饰影响HNF1A转录活性[34]。本文使用常规Western 印迹方法和Far-WB方法对免疫沉淀后的HNF1A进行了O-GlcNAc修饰检测,结果显示,Far-WB结果与Western 印迹一致,进一步验证了方法的可靠性(Fig.5C)。
3 讨论
本文报道了一种O-GlcNAc蛋白快速检测方法。该方法主要基于CpOGAD298N对O-GlcNAc蛋白的广泛识别能力,并结合核心链霉亲和素Stv13蛋白质与生物素之间的特异性结合,将CpOGAD298N与Stv13进行了融合表达。
为了验证方法的实用性,本文基于sOGT蛋白自身O-GlcNAc修饰,制备了阳性对照MBP-sOGT蛋白质。通过活性CpOGA去除O-GlcNAc的功能,制备了去O-GlcNAc修饰的阴性对照MBP-sOGT-DG蛋白质,并通过常规Western 印迹方法对两种对照的糖基化进行了验证,证明了对照的可靠性。
进而通过CpOGAD298N-Stv13融合蛋白质和商业化biotin-HRP建立了Far-WB 检测方法,并通过对照蛋白质、HEK293T细胞总蛋白质及HNF1A蛋白质对新方法进行了验证。本方法全部检测过程可在5-7 h内完成,优于已知基于GST-CpOGAD298N的Far-WB方法。
综上,我们对已有Far-WB检测方法进行了优化,报道了一种更为高效的蛋白质O-GlcNAc修饰检测工具。
