溶酶体半乳糖脑苷酯酶的作用机制及疾病
2024-04-23尹秋媛孟明耀孙建伟
沈 畅, 尹秋媛, 孟明耀, 孙建伟*
(1)云南大学生命科学中心,云南省细胞代谢与疾病重点实验室,省部共建云南生物资源保护与利用国家重点实验室,生命科学学院, 昆明 650500,2) 昆明市延安医院,云南省肿瘤免疫防治研究重点实验室,昆明 650051)
溶酶体贮积症(lysosomal storage diseases, LSD)是一类遗传性代谢疾病,是由于基因突变导致溶酶体中有关酸性水解酶缺陷,进而机体中相应的生物大分子不能正常降解并在溶酶体中贮积,引起细胞组织器官功能的障碍。神经鞘脂贮积症是溶酶体贮积症中的一种,指鞘脂降解所需的溶酶体酸性水解酶缺陷或缺少了神经鞘脂激活蛋白质,造成了不同的鞘脂例如脑苷脂、神经节苷脂或鞘磷脂在溶酶体中贮积,引起中枢神经系统及其他组织病变[1-3]。
β-半乳糖脑苷酯酶(galactocerebrosidase, GALC)是一种溶酶体的酶,参与催化去除β-半乳糖脑苷脂(也称作β-半乳糖神经酰胺) 中的β-半乳糖,从而获得游离神经酰胺 (ceramide)。髓鞘中的鞘脂底物半乳糖神经酰胺(galactosylceramide,GalCer) 是GALC最主要的底物[4, 5]。
GALC功能的异常导致许多重大疾病的发生。人类GALC基因的缺乏会导致克拉伯病(Krabbe disease, KD), 是一种常染色体隐性遗传的鞘脂病,其特征是由于神经毒性代谢产物鞘氨醇半乳糖苷的积累导致少突胶质细胞变性和进行性脱髓鞘[4, 6]。迄今已发现130多种GALC编码基因突变,其中至少有128种突变导致克拉伯病[7]。此外,GALC在肿瘤进展过程中扮演着双重角色,一方面GALC可能是抑癌因子,GALC的下调会导致通过 GalCer水解产生的神经酰胺水平下降,从而促进肿瘤发展[8, 9];另一方面,GALC也可能以原癌基因的作用方式影响肿瘤和肿瘤基质成分,从而参与癌症发展[10]。研究发现,GALC活性的丧失会严重影响血管生成过程、改变癌症相关成纤维细胞的活性,二者均为肿瘤进展过程中的重要参与者[10]。最后,GALC还与阿尔茨海默病、帕金森氏病、多发性硬化症等多种神经退行性疾病有关[11-13]。这些突变背后的生物学作用机制仍有待进一步深入研究。
GALC可能成为诊断、预防和/或阻断溶酶体贮积症、多种癌症及神经退行性病变的潜在靶点。因此,研究GALC在疾病中的作用机制,将为研发相关治疗药物及疗法提供重要借鉴。
1 β-半乳糖脑苷酯酶基因结构
人类GALC基因位于14号染色体的第31位 (14q31),由17个外显子组成,编码669个氨基酸[14, 15],小鼠GALC蛋白与人类GALC蛋白有83%的相似性,斑马鱼中表达2个GALC同源物(名为galca和galcb),与人类和小鼠基因具有相同的17个外显子/16个内含子结构[16]。斑马鱼Galca和Galcb氨基酸序列与哺乳动物GALC的序列相似度为61%,系统发生树分析表明,脊椎动物GALC在进化过程中高度保守[15, 16]。
人类GALC基因的非翻译5′,侧翼区-176至-24富含GC,它包括潜在的YY1(AAATGG)和Sp1(CCCGCC)结合位点,并具有启动子活性。然而与其他溶酶体酶启动子不同的是,人类GALC基因的最小启动子区域不包含看家基因典型的GC-框,也不包含TATA或CAAT 框类型。此外,内含子1的5′端包含6个潜在的Sp1结合位点、1个AP1结合位点和8个AP2结合位点[15, 17]。
2 β-半乳糖脑苷酯酶及酶属性
溶酶体是细胞内重要的物质降解中心,其pH值由ATP依赖性质子泵维持在 4.5~5.0之间,并含有大约60种不同的水解酶,包括硫酸酯酶、糖苷酶、肽酶、磷酸酶、脂酶以及核酸酶,可分解包括糖胺聚糖、鞘脂、糖原和蛋白质等多种底物[18]。缺乏一种溶酶体水解酶将导致无法降解该酶的相应底物,从而导致特定的溶酶体贮积症(lysosomal storage diseases, LSDs)。近三分之一的溶酶体水解酶参与脂质代谢,其中有9种专门降解鞘脂。鞘磷脂的极性头基由不同的8种水解酶裂解,而酸性神经酰胺酶则将最终的神经酰胺骨架裂解为鞘氨醇[19](Fig.1)。
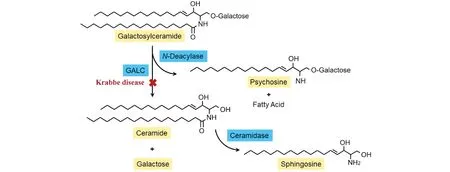
Fig.1 The reaction catalyzed by β-galactocerebrosidase (GALC) and the direct conversion of galactoserebrosides to psychosine when GALC is deficient Acid ceramidase then cleaves the final ceramide backbone to sphingosine
β-半乳糖脑苷酯酶由人类的GALC基因编码,是一种定位于溶酶体的酶,参与碳水化合物在溶酶体中降解加工的最后一步反应,催化水解半乳糖神经酰胺、鞘氨醇半乳糖苷、乳糖甘油酰胺和单半乳糖甘油酯的半乳糖酯键,进而从底物中去除半乳糖,获得游离神经酰胺 (ceramide)。其中,髓鞘中的鞘脂底物半乳糖神经酰胺(galactosylceramide,GalCer)是GALC最主要的底物[20](Fig.1)。
人类GALC蛋白有6个潜在的天冬酰胺N型糖基化位点。前体GALC在内质网中合成为80 kD的前体蛋白质,具有3个结构域,随后在内质网-高尔基复合体中产生并糖基化修饰,GALC前体酶通过甘露糖-6-磷酸(mannose 6-phosphate, M6P)途径被运送到溶酶体。与许多溶酶体水解酶一样,它在溶酶体中被裂解为2个亚基,一个是50 kD的亚基,另一个是30 kD的亚基[15, 21, 22]。但GALC蛋白裂解的功能意义尚不清楚。体外研究表明,前体形式可能具有酶活性,但GALC功能对pH值的要求意味着大多数GALC活性可能发生在溶酶体中,而裂解形式在溶酶体中占主导地位。与其他鞘脂酶一样,GALC也与非酶性鞘磷脂激活剂蛋白皂苷 A(saponin A)联合作用并发挥其水解活性[23]。M6P 标记的GALC也可能进入另一种分泌途径,在细胞外环境中释放。一旦进入细胞外环境,GALC通过 M6PR依赖性途径被回收并输送到溶酶体[21]。据推测,细胞外GALC的再吸收也可能是通过M6P依赖性机制进行的,这可能是由于其蛋白质结构中存在凝集素结构域的缘故[24]。
为研究GALC基因的功能,Suzuki和Wenger研究团队同时尝试GALC蛋白的提取,但从组织中提纯GALC蛋白非常困难,一是由于GALC蛋白在各个组织的表达量非常低,二是由于GALC的强疏水特性[25]。1993年,经过多年尝试的Wenger研究团队从人类尿液中成功纯化了GALC蛋白[25, 26]。随着GALC cDNA和基因的分离和测序[15],使得由GALC突变所导致的疾病研究越来越多,尤其是一种先天性代谢疾病,球形细胞脑白质营养不良症(globoid cell leukodystrophy, GLD),也称作克拉伯病[25], 此外,后续越来越多的研究发现,GALC基因的突变还与帕金森氏病及多种癌症的发生发展有着密切的关系。
3 β-半乳糖脑苷酯酶结构及相关致病突变与疾病
正是由于GALC蛋白的重要作用使得对其蛋白质三维结构的解析显得尤为重要。目前,小鼠GalC蛋白的晶体结构已得到解析(PDB: 3ZR5; in complex with d-galactose: 3ZR6),由结构显示,GALC是碳水化合物活性酶(carbohydrate-active enzymes, CAZy)糖苷水解酶家族59(glycoside hydrolase family 59, GH59)的代表成员[24]。整体三维结构包括3个结构域:中央的三磷酸酯异构酶(triosephosphate isomerase, TIM)桶,β-三明治结构域和凝集素结构域[24]。其中,天冬酰胺残基N284, N363, N387, N542,N585 和N629是潜在的糖基化位点(Fig.2)。为进一步研究其底物结合特性,GALC与D型半乳糖(D-galactose)的共结晶结果表明,与其他含有TIM桶的酶相似,GALC结合口袋的核心是由TIM桶C端的长环形成的,同时β-三明治结构域和凝集素结构域的环也有助于底物结合口袋的形成[24]。

Fig.2 Two orthogonal views of the GALC structure The ribbon diagrams are colored by structural domains: β-sandwich (red), TIM barrel (blue), linker part (orange) and lectin structural domain (green). Disulfide bonds (yellow spheres), calcium ions (gray spheres), and glycosylated molecules (purple rods) are shown. The N-terminus and the C-terminus are marked with labeled circles (Top)[24]
GALC蛋白结构的解析为深入了解GALC基因突变相关疾病的发病机制提供了重要线索。GALC基因的70多个突变与严重的临床表型有关,这些突变在整个GALC序列中均有分布[27-29]。在GALC基因中已经发现了影响基因剪接和mRNA稳定性的突变,或导致缺失、移码突变和错义突变[27-31]。其中,导致克拉伯病的错义突变中,近70%的错义突变涉及酶内部结构中残基的修饰[24],有较大一部分可能导致蛋白质错误靶向或过早降解[32]。错误靶向可能发生在GALC加工途径的几个阶段,包括由于错误折叠而无法从内质网转运到高尔基体,以及由于与M6P受体的结合发生改变而阻碍了GALC向溶酶体的转运。另外,也有研究表明,造成GALC的错误折叠或不稳定的突变并不一定会导致酶活性的完全丧失,仍保留了部分酶活性,这些导致严重错误折叠的突变包括TIM桶中的E114K和S257F,β-夹层结构中的L364R和W410G,以及凝集素结构中的G537R和L629R。再者,如果GALC上的突变干扰了其与溶酶体中激活因子(包括苷类,saposins)的结合,尽管GALC可以被正确加工并定位到溶酶体,但确无法获得底物来进行有效的裂解[32]。
在内质网-高尔基系统中,GALC通过在特定的天冬酰胺残基上添加N型糖基化修饰,对GALC的正确运输与M6P受体的结合至关重要[33]。除6个已知的天冬酰胺残基外,在阿拉伯血统的克拉伯病患者中发现的一个较为常见的突变,D528N,D528位于凝集素结构域的1个环上,这是一个新的糖基化位点,突变的GALC蛋白高糖基化,不能被细胞有效吸收,也不能在溶酶体中发挥功能[30, 32]。
一些致病突变不影响GALC的酶活性、加工或定位,但可能干扰与激活因子的结合。一项研究表明,疾病相关突变E215K对GALC酶活性有较温和的影响,从结构上显示,该位点暴露在TIM桶的表面,带了相反的电荷,干扰了与激活因子的结合[34]。在克拉伯病中,GALC表面非常接近底物结合袋的残基P302突变为精氨酸,这一突变将显著改变该区域的表面特性。另外,β-三明治结构域的长环区域是底物结合部位的一个组成部分,这个环顶端的R380直接与活性部位的半乳糖分子结合,R380W或R380L的突变导致严重的婴儿克拉伯病[24]。越来越多的研究发现GALC的致病突变,因此,对结构的解析为进一步了解克拉伯病和开发潜在的治疗方法提供了重要的借鉴。
3.1 GALC与克拉伯病
1916年,克拉伯病由Knud H. Krabbe博士首次发现,也称作球形细胞脑白质营养不良症,并从组织学水平进行了详细描述[35]。克拉伯病是一种常染色体隐性疾病,由GALC基因突变引起,导致溶酶体水解酶、β-半乳糖脑苷酯酶的活性降低。其病理特点是进行性的严重脱髓鞘(demyelination),根据发病年龄和症状的严重程度分为3个亚型:婴儿型(症状在3岁前出现;一些医生和研究人员进一步将其分为早期婴儿型和晚期婴儿型),少年型(3岁后发病),以及成人型(18岁后发病)。其中婴儿型是为最常见,占大约90%。在婴儿期发病的儿童,在出生时一般表现正常,但出生后的头6个月里,症状迅速发展,出现视力丧失和严重的运动功能退化,患儿通常在2至4岁时死亡[25]。据估计,在欧洲的主要地区,其发病率约为每17万名新生儿中有1人,但在遗传同质化的群体中,特别是在德鲁兹和阿拉伯人中,其发病率可能高达每170名活产儿中有1人。迄今为止,人类基因突变数据库(HGMD)已收录了 130 多种GALC突变。据报道,其中至少有 128 种突变可导致克拉伯病(http://www.hgmd.org)[6, 7]。
GALC负责从含有半乳糖的鞘脂(例如半乳糖甘油酯)中分解去除半乳糖。GALC的缺乏使身体无法代谢一些半乳糖鞘脂,从而在体内不断累积。GALC在大多数组织中均有表达,但大多数疾病过程发生在神经系统中。从组织学水平显示,大脑白质是病变的主要部位,含有“巨大的多核胶质细胞”,这就是现所称的球状细胞(globoid cells)。研究发现一位三岁病逝患儿的脑中,半乳糖神经酰胺(GalCer)在球状细胞中显著升高[36]。1972年Miyatake和Suzuki发现,GALC的底物之一即鞘氨醇半乳糖苷(galactosylsphingosine),通常被称为神经氨酸(psychosine),在正常人脑中,神经氨酸的量非常低,而在克拉伯病患者的大脑中,尤其是在白质和外周神经中积累,高浓度的神经氨酸具有细胞毒性,并引发一连串的致病机制,主要是通过破坏细胞膜以及激活细胞凋亡途径[37]。神经氨酸的积累已在KD患者、KD的Twitcher小鼠模型、基因编辑小鼠KD模型以及KD的犬模型中得到证实[38, 39]。由此提出了“神经氨酸假说”,即克拉伯病的临床表现是由于神经氨酸在中枢神经系统和周围神经系统的积累,该假说认为,由于代谢障碍阻止了半乳糖神经酰胺向半乳糖和神经酰胺的正常分解,半乳糖神经酰胺转而被脱酰基为神经氨酸,而神经氨酸由于GALC的缺乏而不能被分解[40]。
克拉伯病中GALC基因的突变造成了神经系统各个水平的病变,包括神经炎症、轴突病变及突触并发症等[6, 11, 41]。其中,神经炎症是KD的一个主要特征,KD 表型最早的临床表现之一就是不明原因的发热,这表明先天性免疫反应释放了致热细胞因子[42]。患者和所有动物模型的中枢神经系统都表现出强大的星形胶质细胞增生、小胶质细胞活化和巨噬细胞招募[42, 43]。最近对早期病理进行的研究表明,神经炎症先于髓鞘的变化或缺失,但炎症的诱因仍需进一步研究。在一些含有致病的GALC突变基因的小鼠品系中,包括twitcher (W332X), twi-5J (E130K), 以及twitrs,它们都在终末阶段表现出明显的神经炎症,包括小胶质细胞和星形胶质细胞活化以及巨噬细胞浸润[40, 44, 45]。研究发现,早在出生后2周,twi-5J 小鼠的前脑中就出现了反应性小胶质细胞(reactive microglia)和星形胶质细胞,胶质纤维酸性蛋白GFAP (glial fibrillary acidic protein)免疫反应逐渐加强,3周时小胶质细胞形成了离散的结节,周围是肥大的星形胶质细胞[46]。5周时几乎所有的小胶质细胞形态均异常。在这个过程中,许多细胞因子和趋化因子的转录水平随之增加,包括TLR1 (Toll-like receptor 1)、TLR2 (Toll-like receptor 2)、Ccl3(C-C motif chemokine 3)、Ccl5 (C-C motif chemokine 5)、Cxcl10 (C-X-C motif chemokine 10)、IL-6 (interleukin-6) 及TNF-α ( tumor necrosis factor α)等。但导致炎症产生的具体机制仍需进一步研究[46]。
对鞘氨醇半乳糖苷致病机制的研究发现,GALC缺陷会产生内质网应激、氧化应激、代谢紊乱或错误折叠蛋白质的产生等变化,从而导致细胞膜动态紊乱(特别是在脂质筏内)、信号转导途径紊乱、蛋白体降解途径过度激活并促进细胞凋亡[47]。一方面,GALC功能失调导致髓鞘膜内鞘氨醇半乳糖苷累积,髓鞘膜内脂质成分的紊乱会影响细胞表面受体或膜相关信号分子的正常表达,而当通过向培养细胞中注入外源性鞘氨醇半乳糖苷,发现加入少于10 mM的鞘氨醇半乳糖苷不会产生任何变化,但浓度较高时则会产生许多影响,包括细胞凋亡、干扰鞘氨醇-1-磷酸信号传导;以及蛋白激酶 C (protein kinase C, PKC)、肿瘤坏死因子(tumor necrosis factor, TNF)、白细胞介素-6 (interleukin-6, IL-6)、诱导型一氧化氮合酶 (inducible nitric oxide synthase, iNOS)、磷脂酰肌醇 3-激酶 (phosphoinositide 3-kinase, PI3K)、前列腺素 D2 (prostaglandin D2, PGD2)的表达或功能的变化[42, 48]。另一方面,外源性鞘氨醇半乳糖苷还会影响线粒体功能,破坏电子传递链、线粒体膜电位和过氧物酶体[48-51]。同时,在少突胶质细胞和雪旺细胞等髓鞘细胞内,鞘氨醇半乳糖苷会在溶酶体和脂伐的微区域累积。因此,溶酶体-内吞体通路、内吞和膜受体介导的信号传导会随着鞘氨醇半乳糖苷水平的增加而逐渐中断,例如参与不同信号级联的PKC激酶的蛋白质水平降低,干扰少突胶质细胞内髓鞘基因的表达[52]。以上是病理过程的累积所造成的结果,那么内源性鞘氨醇半乳糖苷在细胞内有何作用,研究表明,内源性鞘氨醇半乳糖苷的积累可激活磷脂酶 A2 (phospholipase A2, PLA2),通过生成生物活性脂质溶血磷脂酰胆碱和花生四烯酸,激活细胞死亡信号级联或生成活性氧[53, 54]。此外,溶酶体功能障碍导致的鞘脂代谢变化也会改变鞘脂代谢物的水平,例如神经酰胺、神经酰胺-1-磷酸和鞘磷脂-1-磷酸,它们是炎症中的重要信号分子[55]。但要全面深入的了解内源性鞘氨醇半乳糖苷的作用机制,仍需要对其调节的细胞通路进行更多研究 (Fig.3)。

Fig.3 Mechanisms of Krabbe disease Mutations in the GALC gene cause loss of function of the encoded protein β-galactocerebrosidase, resulting in the accumulation of psychosine. High levels of psychosine are cytotoxic, leading not only to disturbances in the dynamic structure of the cell membrane, but also to a gradual disruption of endosomal-lysosomal signaling with increasing levels of psychosine, as well as varying degrees of disruption of the mitochondrial function, ultimately leading to progressive neuronal demyelination
综上,GALC基因突变引起的克拉伯病,造成了神经系统各个水平的病变,深入的机制研究揭示GALC缺陷会造成内质网应激、氧化应激、代谢紊乱或错误折叠蛋白质的积累等后果,从而导致细胞膜动态、信号转导途径紊乱以及蛋白酶体降解途径过度激活、最终促进细胞凋亡。
3.2 GALC与神经退行性疾病
如前所述,GALC在溶酶体内发挥作用,将β-半乳糖从半乳糖神经酰胺(GalCer)和神经氨酸去除,从而产生神经酰胺 (ceramide)和鞘磷脂。神经酰胺是所有复合鞘磷脂的核心成分,在细胞的许多基本过程中发挥着重要作用,包括生长、分化、细胞周期停滞、衰老、存活和凋亡。研究证实,神经酰胺参与了阿尔茨海默病的神经细胞死亡过程。正常情况下,神经酰胺的合成受到了严格的调控,但在神经退行性疾病发生时,其合成就发生了紊乱[11-13, 56]。
一项研究测定了40份人脑标本中6种神经酰胺的含量,发现在有多种神经病理学异常的样本中,神经酰胺的含量显著增加。另外,与同年龄的正常对照组相比,在神经病理学异常患者标本中,神经细胞中调控神经酰胺水平4个基因GALC、鞘磷脂磷酸二酯酶(sphingomyelin phosphodiesterase, SMPD1)、鞘磷脂磷酸二酯酶 3 (sphingomyelin phosphodiesterase 3, SMPD3)的表达量显著上调,而神经酰胺葡萄糖基转移酶(ceramide glucosyltransferase, UGCG)的表达量呈下调趋势[56]。
近些年,利用全基因组关联研究发现了多种疾病的遗传风险因素,最近的一项帕金森氏病的全基因组关联分析研究发现,GALC基因座上的rs979812 变异与半乳糖甘油酯酶活性增加有关,半乳糖基甘油三酯酶活性的增加可能与帕金森病存在因果关系[12]。此外,GALC基因的变异也被确定为多发性硬化症(multiple sclerosis, MS)的风险因素之一,但这些变异与多发性硬化症的生物学相关性仍不完全清楚[13]。这些发现表明,GALC可能成为诊断、预防和/或阻断神经退行性病变过程的潜在靶点。
3.3 GALC与癌症
GALC蛋白的催化产物神经酰胺在肿瘤中充当着抑癌代谢物的重要角色,神经酰胺代谢的变化不仅会促进肿瘤细胞的存活,还会对化疗产生抗性。由于神经酰胺处于各种相互关联的代谢途径的中心,细胞内神经酰胺的水平和定位是许多鞘脂质代谢酶协调作用的结果。GALC是催化产生神经酰胺的酶之一,近些年的研究表明GALC在肿瘤生长和分化过程中发挥了双重作用,某些癌症类型中充当抑癌因子,而另外一些癌症中则发挥致癌因子的作用[8]。因此,对GALC在癌症中的具体作用机制还有待深入研究。
3.3.1 GALC作为抑癌因子 溶酶体GALC能从β-半乳糖基神经酰胺中去除β-半乳糖,从而生成抑癌代谢物神经酰胺。近些年,大量的研究证据表明,鞘磷脂和参与其代谢的酶在肿瘤细胞的生物学中发挥着关键作用。喉鳞状细胞癌、头颈部肿瘤以及鼻咽癌中都存在与启动子高甲基化相关的GALC下调现象,在鼻咽癌中,低水平的GALC免疫反应与较高的淋巴结转移风险有关[8]。这些数据表明,GALC 可能是一种抑癌的酶,它的下调会导致神经酰胺水平下降,从而使肿瘤发展。此外,GALC 活性的降低也可能导致其底物GalCer水平的升高。与该研究结果相似的是,GalCer合成酶UDP神经酰胺:半乳糖的半乳糖基转移酶(UDP-galactose:ceramide galactosyltransferase, CGalT) 的上调,有助于提高基底样乳腺癌的侵袭性[57](Fig.4)。

Fig.4 GALC acts as both oncosuppressor enzyme and oncogenic enzymes in cancers Representative examples of GALC in different cancer types are shown
3.3.2 GALC作为致癌因子 尽管已有结果表明,GALC可能发挥癌症发展的抑制作用,但最近的数据表明,GALC也可能以原癌基因的作用方式影响肿瘤和肿瘤基质成分,从而参与癌症发展。第一个证据来自于对twitcher小鼠模型(上述提及的克拉伯病模型)的观察,发现GALC活性的丧失会严重影响血管生成过程,而血管生成过程是癌症的标志[10]。GALC还可能改变癌症相关成纤维细胞的活性,这些成纤维细胞是肿瘤进展过程中的重要参与者。事实上,过表达GALC的成纤维细胞会刺激结直肠癌细胞的增殖、迁移和致瘤潜能[58]。但GALC导致这些后果的具体作用机制目前仍未定论,研究者们提出了一些猜测,一方面GALC 高表达的细胞可能会释放原癌介质,另一方面细胞外M6P修饰的GALC蛋白可能会与M6P/胰岛素样生长因子2受体(insulin like growth factor 2 receptor, IGF2R)相互作用,从而产生“旁分泌”的活性,最终刺激结直肠癌细胞的增殖、迁移和致瘤潜能[59]。
除了影响肿瘤基质成分,GALC还会改变癌细胞的行为。据报道,高水平的GALC免疫反应与结直肠癌患者的不良预后有关,循环肿瘤细胞中的GALC表达水平与非小细胞肺癌患者的远端转移、肿瘤数量和对治疗的不良反应密切相关[58, 60]。此外,基因干扰GALC会导致宫颈癌HeLa-S3细胞的细胞周期异常,表现为细胞分裂中期阶段延迟以及细胞分裂失败[61]。与这些观察结果一致的是,GALC敲降抑制小鼠和人类黑色素瘤细胞的致瘤和转移活性[62]。在黑色素瘤细胞中,GALC敲降细胞的鞘磷脂水平异常,细胞内抑癌代谢物神经酰胺的水平升高,同时鞘磷脂磷酸二酯酶3 显著上调。SMPD3催化鞘磷脂水解形成神经酰胺和磷酸胆碱。与这些发现相一致的是,在从普通痣到IV期人类黑色素瘤的癌症进展过程中,GALC表达的逐渐增加与SMPD3蛋白和神经酰胺免疫反应的水平成反比[62]。SMPD3表达量上升会影响黑色素瘤的生长和对免疫疗法的反应[63]。因此,SMPD3的上调造成细胞内神经酰胺生成的增加,可能会介导黑色素瘤细胞中GALC下调所产生的抑制作用。Sp1和Sp3转录因子、p53和TNFα等多种转录因子均可调控SMPD3的转录[10]。因此要阐明黑色素瘤细胞中GALC和SMPD3表达之间存在的负相关性的分子决定因素,仍需要进一步的研究[64]。
值得注意的是,在人类黑色素瘤中,与内质网/高尔基细胞成分相关基因本体术语(GO terms)中,前25个最显著的基因与GALC表达量呈正相关。而编码参与鞘脂代谢的其他溶酶体酶的基因却无该相关性,这些基因在黑色素瘤中的表达与富含溶酶体细胞成分相关基因语义注释的基因集有关。这些研究结果提出了一个未回答的疑问:GALC是否可能参与调节影响肿瘤进展过程中的内质网/高尔基细胞内途径,例如自噬和内质网应激,并影响癌细胞中内质网膜/高尔基相关SMPD3的水平[64](Fig.4)。
综上所述,GALC 在肿瘤进展过程中扮演着双重角色。因此,要剖析GALC对癌症的影响仍需进一步深入的研究。例如,关于GALC调控的肿瘤和基质细胞脂质组、转录物组、蛋白质组和代谢组的变化,目前几乎未见任何数据。再者,人们普遍认为GALC只是一种“鞘氨醇半乳糖苷清除剂”,并未考虑到这种酶在细胞生物学中所产生的更广泛影响。同样,在肿瘤进展和转移过程中调节GALC表达的机制也是未知的。因此,了解癌基因和癌基因抑制子的突变是否会影响,以及如何影响不同肿瘤类型中GALC的表达和活性至关重要[64]。
鞘磷脂和参与其代谢的酶在肿瘤细胞的生物学中发挥着关键作用,那么,影响抗肿瘤代谢物神经酰胺水平的酶会作为潜在的肿瘤促进或抑制的靶点,更好地研究这类酶在肿瘤细胞行为和对治疗的反应中的作用,这将会为癌症的新型治疗策略提供借鉴[65]。
4 β-半乳糖脑苷酯酶相关疾病的治疗
GALC负责分解某些糖脂,包括有毒化合物鞘氨醇半乳糖苷。GALC相关疾病的组织学表型包括中枢和周围神经系统髓鞘脱落、轴突变性、胶质细胞增多、神经炎症和少突胶质细胞死亡,克拉伯病患者还表现出神经功能的衰退[6, 11, 41]。
尽管克拉伯病是一种单基因疾病,但由于它的生化、组织学和临床表型非常复杂,可能的治疗目标也较多。研究者们已经利用多种GALC突变的模型,Twitcher小鼠、犬类模型、灵长类模型等开展了一系列治疗的研究,这些研究虽然为了解疾病发生发展提供了重要线索,但利用其开发治疗手段收效甚微。目前,针对GALC缺乏造成的克拉伯病的治疗,主要通过单一模式疗法(single-modality therapies)及多模式疗法(multi-modality therapies)[66, 67]。
克拉伯病的单一模式疗法主要包括以下9类,骨髓移植(bone marrow transplant)、酶替代疗法 (enzyme replacement therapy)、病毒介导的基因疗法(viral-mediated gene-therapy)、底物减少疗法(L-环丝氨酸)(substrate reduction therapy (L-cycloserine))、抗炎疗法(anti-inflammatory therapies)、抗氧化疗法(antioxidant therapy)、少突胶质细胞移植(oligodendrocyte transplant)、神经元和间充质干细胞移植(neuronal and mesenchymal stem cell transplant)、神经干细胞基因疗法(neural stem-cell gene therapy)。纠正功能性GALC的缺乏是最直接的治疗方法,可以通过酶替代或基因治疗直接向中枢神经系统提供活性酶,或者移植表达GALC的供体细胞。另一种可行的策略是通过抑制鞘氨醇半乳糖苷的合成(底物减少疗法)来减少其累积。针对继发性致病机制(例如神经炎症和氧化应激)的治疗也能用于KD的治疗,但这些策略并不能完全纠正疾病,因为酶缺乏的问题并未得到解决[66, 67]。
在临床前试验中,目前最有效的方法是多种致病机制/途径的综合治疗,也就是上述方法组合的多模式疗法。目前,KD患者的标准治疗方法是造血干细胞移植(hematopoietic stem cell transplantation, HSCT),以及与造血干细胞移植同时进行的各种联合疗法[66]。
造血干细胞移植来源于骨髓(bone marrow transplant, BMT)或脐带血(umbilical cord blood, UCB)。使用造血干细胞移植治疗婴幼儿KD患者的成功率不高,但可以有效治疗青少年、幼年或成年发病者。据推测,造血来源的GALC阳性供体细胞可迁移到中枢神经系统,并提供持续的低水平的GALC。供体细胞分泌的GALC分别通过网状内皮系统细胞每个细胞表面的甘露糖-6-磷酸(M6P),或甘露糖受体被中枢神经系统中的缺陷细胞吸收。与细胞表面受体结合后,GALC被内吞并通过内体系统转运至溶酶体,在溶酶体中与受体分离。这一过程被称为交叉校正(cross-correction),能够为中枢神经系统的细胞提供功能性的GALC。例如,已有研究表明,将来自Twitcher小鼠的少突胶质细胞移植到缺乏髓鞘碱性蛋白的Shiverer小鼠体内,它们能够存活并正常髓鞘化轴突。这些研究数据表明,移植的少突胶质细胞吸收了宿主细胞(shiverer)中足够的GALC 活性,从而纠正了代谢缺陷。同时也有研究发现,造血干细胞移植能降低疾病的严重程度,包括减少癫痫发作活动、提高运动能力和语言发育[66, 67]。
由于疾病表现和发病机制的复杂性,迄今为止,单一模式疗法对治疗克拉伯病的效果微乎其微。针对多种致病机制的疗法不仅能延长患者的寿命,还能减少发育不良的情况。但要开发出真正有效的治疗方法,仍需解决有关KD发病机制的基本问题,因此,对于GALC基因的功能研究显得尤为重要。
5 问题与展望
在溶酶体贮积症及其相关疾病领域,GALC基因及其编码的β-半乳糖脑苷酯酶成为了其中一个研究焦点。其在多种疾病包括克拉伯病、多种癌症、神经退行性疾病等中的作用,揭示了其在细胞生物学和病理学中的重要地位。然而,关于GALC基因突变背后的具体生物学作用机制,尤其是在不同疾病背景下的多样性和复杂性,仍然是一个待解决的科学问题。
在将来的研究中,GALC基因及其编码酶在疾病发生、发展中的角色需要进一步的深入研究。首先,对GALC基因的多种突变形式及其在不同疾病中的具体作用机制进行深入探讨,将有助于我们更全面地理解其在疾病中的功能。其次,GALC在不同类型的细胞和组织中可能存在不同的功能和作用路径,这些差异如何影响疾病进程,值得进一步探索。在此基础上GALC及其相关通路有可能成为未来疾病治疗的潜在靶点。开发针对GALC的小分子药物、基因治疗策略或利用CRISPR等基因编辑技术修复GALC基因突变,均有可能成为未来治疗相关疾病的有效手段。同时,GALC也可能用于疾病的早期诊断和预后评估,为临床提供更多的治疗选择和决策依据。总之,GALC在多种疾病中的作用及其作为潜在的治疗靶点的研究,将为未来的生物医学研究和临床治疗提供新的方向和可能性。
