基因编辑技术及其在糖生物学研究中的应用
2024-04-23章恩华
章恩华, 邱 宏,*
(1)中国科学院上海药物研究所, 上海 201203;2)南京中医药大学新中药学院, 南京 210023)
Capecchi等人在上个世纪80年代发展了基于同源修复的基因打靶技术,用于基因功能的研究,但是该技术靶向整合效率低、筛选耗时费力、随机突变风险高而且应用场景受限[1-3]。学者们尝试了不同方法改进同源重组的效率,但是并未获得显著进步。归巢核酸内切酶(meganuclease)的功能研究发现,双链DNA切口(double strand breaks,DSBs)的形成可以显著提高同源重组修复效率(50~1 000倍)[4, 5]。这一发现促进了能够识别特异性核酸序列的核酸酶嵌合体的开发和应用[6]。锌指核酸酶(zinc-finger nucleases, ZFNs)是最早被成功应用于基因编辑的核酸酶嵌合体[7, 8],随后转录激活样效应因子核酸酶(transcription activator-like effector nuclease, TALENs)也被成功用于基因编辑[9, 10]。它们主要通过构建能特异性识别基因组DNA序列的核酸内切酶FokI嵌合体,在特定位置产生DSB,再通过同源重组(homologous recombination,HR)的方式导入外源基因,或者通过非同源末端修复(non-homologous end joining,NHEJ)连接断裂的DNA,利用随机插入/删除(insertion &deletion , indel)引起移码突变敲除靶基因[11]。但具备DNA序列特异性识别功能的核酸酶嵌合体的筛选耗时费力,而且特异性不够,容易导致随机突变。2012年前后,可以通过互补RNA导向序列实现基因组几乎任意位点的切割的簇集有规律间隔短回文序列-及其相关蛋白( clustered regularly interspaced short palindromic repeats-CRISPR-associated proteins, CRISPR-Cas ) 系统被发现,这一技术规避了复杂的核酸酶嵌合体构建,不仅实现了基因的精准定位,而且显著降低了设计难度和应用成本,该发现被迅速地应用于微生物、植物、动物及人类基因的编辑[12, 13](Fig.1)。最近报道了来源于细菌的水解型内切核酶(hydrolitic endonuleolytic ribozyme, HYER),该核酶及其工程化改造产物均可实现RNA导向的核酸编辑[14]。

Fig.1 Gene editing tools based on programable nucleases ZFN, TALEN and CRISPR/Cas9 generate DSBs at specific locations in different mechanisms, and then introduce desired gene modification through HDR or insertions/deletions by NHEJ that leads to gene knockout
本文首先简要介绍CRISPR-Cas的组成与分类和工作原理,接着总结了靶向DNA和靶向RNA的各种基于CRISPR-Cas的基因编辑技术的特点和应用。从递送角度评估了细胞外囊泡作为CRISPR-Cas递送载体用于疾病治疗的可能性。最后,总结了基因编辑技术在糖生物学研究中的应用。
1 CRISPR-Cas系统的组成与分类
CRISPR-Cas作为原核生物的获得性免疫系统被发现,并进一步被设计为强大基因组编辑工具,离不开对其独特系统组成的研究。而依据其结构不断完善的分类体系也为其功能的多样性开发奠定基础。
1.1 CRISPR-Cas系统的组成
CRISPR-Cas系统由CRISPR基因序列(CRISPR array)以及Cas基因(CRISPR associated, Cas)两部分组成,CRISPR基因转录的RNA识别和定位外源基因,而Cas基因编码的Cas蛋白则在RNA的引导下对目标基因进行定向编辑。
CRISPR基因序列(CRISPR array)主要由前导序列(leader)、重复序列(repeat)和间隔序列(spacer)组成。前导序列富含AT碱基(A-腺嘌呤,T-胸腺嘧啶),位于CRISPR基因上游,被认为是CRISPR序列的启动子;重复序列为长度约20~50 个碱基且包含5~7 bp回文序列,与间隔序列交替排列,其转录产物可以形成发卡结构,发挥稳定RNA二级结构的作用;间隔序列是细菌俘获的外源DNA序列,用于贮存外源入侵信息,当外源遗传物质再次入侵时,细菌CRISPR-Cas系统会被激活,调集RNA引导的Cas核酸酶,序列特异性地沉默噬菌体和质粒与转座子。间隔序列的排列顺序可以反映细菌被外源核酸感染时间的先后,序列越靠近前导序列端(5′端),感染时间越靠后。
1.2 CRISPR-Cas系统的分类
CRISPR-Cas系统结构复杂,缺乏统一的分类标志物,而且各类新的CRISPR类型不断涌现,CRISPR-Cas的分类体系也在不断地调整,这给CRISPR-Cas系统的稳定分类带来巨大挑战[15]。2011年Makarova等人根据Cas基因的系统进化特征、CRISPR 重复序列的序列信息和组织特征,以及CRISPR-Cas位点的结构特征对CRISPR-Cas系统进行分类[16],经过2015年和2020年的2次更新和完善,目前,将CRISPR系统分为2大类、6个类型以及30多个亚型[17-18]。在该分类体系中,2类CRISPR的主要区别在于Cas蛋白组成及效应模块的差异:第1类由多种Cas蛋白共同组成效应模块,其中部分Cas蛋白与crRNA结合形成编辑复合物,参与干扰噬菌体或外源质粒的入侵,其他的Cas蛋白作为辅助蛋白质协助调节免疫过程,包括I型、Ⅲ型和Ⅳ型;第2类效应模块由单一的多结构域Cas蛋白(如Cas9、Cas12、Cas13)组成,包括Ⅱ型、Ⅴ型和Ⅵ型。
基于Cas蛋白功能的异同,可以将CRISPR-Cas系统中的Cas分为4大功能模块:(1)适应模块:主要作用为识别外源核酸,从中抓取原间隔子序列、将其加工成间隔序列并将间隔序列整合入CRISPR基因序列,主要包括Cas1与Cas2,Cas1-Cas2聚合物几乎存在于所有亚型中,在II-A型中主要为Csn2蛋白。(2)表达处理模块:主要作用为将Pre-crRNA加工处理成crRNA,在第1类中发挥主要催化作用的是Cas6蛋白,第2类Type II主要由RNase III负责,在Type V和Type VI中则由效应Cas核酸酶中一个独立于基因编辑活性的催化中心负责。(3)干扰模块(效应器模块):负责核酸的靶向识别和切割,第1类由多种Cas蛋白质组成,第二类是单一的大蛋白质例如Cas9、Cas12和Cas13。(4)信号传导或辅助模块:包括各种功能已知或未知的辅助蛋白质。
2 CRISPR-Cas系统工作机制
CRISPR-Cas系统通过在CRISPR位点的重复序列之间整合外源核酸片段,从而可以通过CRISPR-Cas系统抵御外源核酸的再次入侵[19]。其发挥免疫防御作用的机制主要分为三个阶段:适应阶段、表达阶段和干扰阶段[18, 20-24]。
2.1 适应阶段
CRISPR-Cas系统加工得到的Cas蛋白复合物可以识别一个独特的原间隔序列邻近基序(protospacer-adjacent Motif,PAM),该序列具有保守性,长度一般为2~5碱基。在与其相邻1~4碱基位置为原间隔序列(protospacer),是噬菌体或是质粒上与间隔序列对应的序列。Cas蛋白通过PAM定位识别并剪切Protospacer,在CRISPR基因序列的5′端重复序列复制完成后,将间隔序列DNA插入到CRISPR基因序列的2个重复序列之间,使其成为新间隔序列。也有一些CRISPR-Cas系统采用了另一种适应机制,即通过在CRISPR-Cas位点编码的逆转录酶的逆转录,从RNA中获得间隔序列。
2.2 表达阶段
CRISPR先被转录成Pre-crRNA(crRNA成熟过程的中间物)再被多个Cas蛋白质形成的复合物或者单一Cas核酸酶不同功能结构域(也有部分为非Cas核糖核酸酶,例如Type Ⅱ中Cas9与RNase Ⅲ共同参与Pre-crRNA的加工)加工处理为成熟crRNA。Type Ⅱ型CRISPR-Cas系统crRNA需先与tracrRNA(反式激活CRISPR相关RNA)互补结合,然后被RNase Ⅲ切割,切割后形成tacrRNA和crRNA末端互补结合的向导RNA,向导RNA可与Cas9核酸酶HNH结构域结合,结合后形成的复合物遂行核酸靶序列的识别和切割功能。
2.3 干扰阶段
成熟的crRNA与Cas核酸酶形成复合物,引导Cas核酸酶识别入侵病毒或者质粒中的protospacer,再被Cas蛋白结构域的功能区裂解和灭活。以TypeⅡ型为例,crRNA与tracrRNA或者sgRNA和Cas9形成的复合物通过识别PAM,精准定位至靶序列,结合后,靶序列区域的DNA双链被解开,形成R-Loop。此时,sgRNA与其中一条DNA链形成互补链,另一条保持游离态。Cas9蛋白随后会精准地于PAM上游3个核苷酸的位置切割核酸,形成平末端产物。Cas9蛋白的HNH结构域识别并切割与sgRNA碱基互补配对的外源DNA链,RuvC结构域识别并切割不与sgRNA互补配对的DNA链。CRISPR-Cas系统就是利用这样的DSB修复机制沉默外源核酸,使其灭活(Fig.2)。
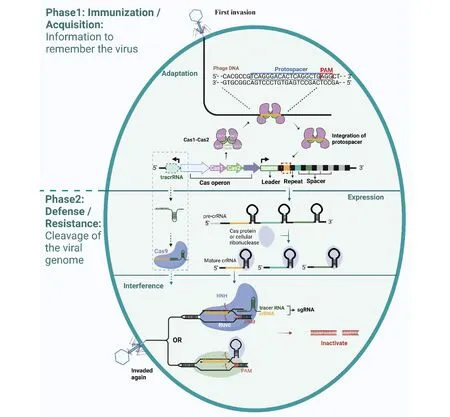
Fig.2 The adaptive immune response based on the CRISPR-Cas system The CRISPR Cas system first catch, select, obtain, and integrate protospacer sequences from invading gene elements (adaption). When the cells undergo a second invasion, the cells would express the CRISPR-associated RNA (crRNA) (expression) and the Cas protein like Cas9 would be directed to the specific DNA loci to generate a DSB (interference)
3 CRISPR-Cas的工程化改造和应用
成熟crRNA通过碱基互补配对与tracrRNA形成RNA二元复合物,该复合物能够引导Cas9蛋白识别并切割与crRNA互补配对的靶DNA,产生DSB,这一识别需要靶DNA上含有PAM序列NGG;将crRNA和tracrRNA二元复合物改造成单链RNA,所得单链RNA仍具备导向作用,因而这一RNA称做单链导向RNA(single guide RNA, sgRNA)[25]。2013年,基于这一发现,仅需sgRNA和Cas9两个元件的新基因编辑系统被成功开发,并被用于哺乳动物细胞的基因编辑[26-28]。这一基于CRISPR-Cas的第一代基因编辑系统存在脱靶和难递送等问题,随后一直朝着小型化、中靶效率高、伤害与脱靶最小化和组织靶向递送等目标进行工程化改造,产生了CRISPRi、CRISPRa、碱基编辑器和先导编辑器等不同工具,不仅提高了基因编辑的精准度和特异性,同时也将CRISPR-Cas9的应用从基因编辑拓展到了基因表达调控、成像和检测与表观遗传修饰等领域(Fig.3)。
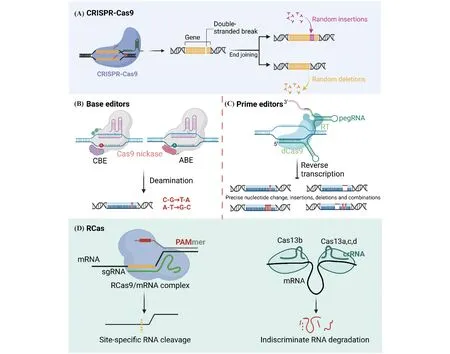
Fig.3 The gene editors based on CRISPR-Cas (A) The CRISPR-Cas9 system introduces indels through DSBs and different cellular DSB repair pathways.(B) CBE and ABE directly generate C-to-T and /or A-to-G transitions without producing DSBs. (C) PE enables insertion, knockout, and substitution of arbitrary small fragments of DNA sequences and significantly reduces off-target rates. (D) The RNA editing system. Cas9 relies on PAMmer for targeted cleavage; the Cas13 system specifically recognizes and targets ssRNAs, and in addition to cleaving paired ssRNAs, it will lead to collateral cleavage on ssRNAs nearby
3.1 基于CRISPR-Cas9系统的基因转录编辑器
2013年2月斯坦福大学亓磊团队发现化脓链球菌Cas9 ( spCas9 ) 的一种缺乏内切酶活性的Cas9(dead Cas9, dCas9)在与gRNA共表达时,产生一种DNA识别复合物,该复合物与DNA靶序列结合后,可以阻断RNA聚合酶或转录因子的结合,干扰转录延伸,将Cas9从RNA引导的核酸酶转化为RNA引导的核酸结合蛋白质[28]。基于dCas9这一特性,亓磊团队开发了基因表达调控工具CRISPRa(CRISPR activation, CRISPRa)和CRISPRi(CRISPR interference, CRISPRi)[29, 30](Fig.4)。
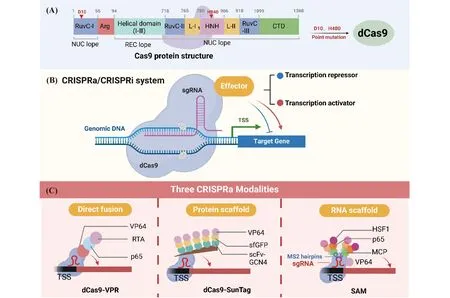
Fig.4 Gene expression regulation based on the CRISPR-dCas9 system (A) Point mutations at both the RuvC domain (D10A) and the HNH domain (H840A) leads to loss of endonuclease activity. The mutated Cas9 was named as dCas9. (B) CRISPRi: The complex formed by dCas9 or dCas9 fused with different transcriptional repression domains with sgRNA downregulates gene transcription by blocking the RNA polymerase from reading through gene sequences or directly inhibiting the expression; CRISPRa: dCas9 is fused to a transcriptional activator domain to activate gene transcription. (C) Three CRISPRa modalities based on different mechanisms: Tandemly fuse different transcriptional activators to dCas9 (dCas9-VRP); fuse a scaffold protein with dCas9 for multiple transcriptional activators binding ( dCas9-SunTag ); simultaneous construction of transcriptional activators fused with dCas9 and sgRNA contains aptamer recognizeing MS2, which can simultaneously bind multiple transcriptional activators (SAM)
3.1.1 CRISPRi系统 在大肠杆菌中,利用一个带有基因靶向序列、Cas9蛋白结合序列和转录终止序列的sgRNA,将dCas9靶向基因的编码序列或其启动子,发现dCas9与sgRNA形成的复合物能以空间位阻的形式阻断RNA聚合酶通读基因序列,直接阻断蛋白质编码区转录的延伸,从而抑制基因转录,这个过程叫做CRISPRi。与RNAi的基因沉默不同,CRISPRi系统不需要破坏转录的mRNA[30]。该系统主要通过2种方式抑制目标基因转录:(1)阻止RNA聚合酶与目标基因的启动子结合抑制转录;(2)与开放阅读框靶向结合,抑制转录延伸(相当于转录终止子)。
在哺乳动物细胞中,上述CRISPRi复合物仅有微弱的基因表达抑制作用[29]。为了提高转录抑制强度,研究者将dCas9与不同转录抑制子融合表达,经筛选发现,dCas9-KRAB融合蛋白具有较高的抑制效率[30]。KRAB(Krüppel-associated box)结构域是锌指蛋白(zinc finger proteins,ZNFs)中的一个高度保守的结构域,长约75个氨基酸,据预测它们可以形成两个双亲性的α-螺旋,目前,大多数CRISPRi研究使用来自KOX1(ZNF 10)蛋白的KRAB,这一结构域也被用于TALEs介导的基因表达抑制[31]。这些KRAB结构域与辅抑制蛋白KAP1(KRAB-associated protein 1, KAP1)结合,而KAP1反过来招募组蛋白甲基转移酶SETDB1(SET domain bifurcated histone lysine methyltransferase 1),促进组蛋白H3K9(H3 lysine 9)的三甲基化,导致转录抑制[32]。最近研究发现,ZIM3 KRAB的基因表达抑制作用强于KOX1 KREB[33]。
3.1.2 CRISPRa系统 通过dCas9与转录抑制调控因子相连构建CRISPR抑制的(CRISPRi)基因表达抑制系统,抑制靶向基因的表达;相应地,将dCas9与转录激活调控因子相连,则能生成一个被称为CRISPRa的转录上调系统。目前的CRISPRa系统可以分为三类[34]:一类是将单个或多个转录激活因子与dCas9串联构建融合蛋白质;第二种是构建dCas9与脚手架蛋白质的融合蛋白质,用以结合多个转录激活因子;第三种是同时构建转录激活因子与dCas9的融合蛋白质和含有MS2适配体(MS2噬菌体的19核苷酸 RNA 发夹结构)序列的sgRNA(用于招募与MS2融合的转录激活因子复合物),这样可以同时结合多个转录激活因子,增强转录激活效应(Fig.4)。3种CRISPRa系统元件均包含dCas9融合蛋白质和sgRNA,区别在于dCas9融合蛋白质和sgRNA的构建方式不同:(1)dCas9融合蛋白质包括①直接融合单个转录激活因子,例如将dCas9的C末端与1个由4个VP16结构域组成的激活结构域VP64融合,在HEK293T细胞中稳定表达时,能够促进转录上调[35, 36];②串联融合多个同种或/和异种转录激活因子;例如筛选发现三元转录激活因子VPR(VP64-p65-Rta)与dCas9的C端融合而得的dCas9-VP64-p65-Rta(dCas9-VPR),激活能力显著增强,其中VP64是单纯疱疹蛋白16的TAD四聚体,p65则是核因子κB (nuclear factor kappa-B, NF-κB) 蛋白质家族中的成员,转录激活蛋白(replication and transcription activator, Rta)是Y疱疹病毒中具有序列保守性的一个蛋白分子[37]。和③通过串联融合与转录激活因子融合蛋白特异性结合的多肽阵列脚手架,例如Sun-Tag[38]。(2)sgRNA包括①原型sgRNA;②可与特定蛋白质标签结合的工程化sgRNA,例如,由dCas9-转录激活因子融合蛋白质、含有与MS2结合的核酸适配体序列的sgRNA和MS2-转录激活因子融合蛋白质三种元件构成的CRISPR-SAM系统(synergistic activation mediator,SAM),能够特异性激活多数细胞内源基因[39]。在SAM系统中,sgRNA含有与MS2结合的适配体序列,可与MS2、p65和HSF1三元融合蛋白质结合,增强转录激活作用,而且启动子区单一位点sgRNA导向的SAM转录激活效果远高于多位点dCas9-VP64的转录激活效果[39]。
3.2 表观基因组编辑器
许多表观遗传修饰因子与dCas9蛋白融合后可在DNA或染色质水平上进行化学修饰,例如通过在DNA上靶向添加甲基或在组蛋白残基上插入乙酰基,以及甲基的表观遗传修饰调节基因表达,这些修饰作用持久并且可遗传[40-43]。构建dCas9表观遗传编辑系统的常用方式是将dCas9与表观遗传修饰酶融合在一起,通过核酸酶靶向结合目标DNA的特性实现特定基因组位点上表观基因组的编辑。
将DNMT3a(DNA methyltransferase 3 alpha),一种能够在体内甲基化CpG位点的活性DNA甲基转移酶)的DNA甲基化结构域与dCas9结合,使靶向启动子上的DNA瞬时甲基化,可以实现长期的基因沉默[44-46]。例如,用dCas-DNMT3a融合蛋白靶向SNCA(synuclein alpha)内含子1,能在携带SNCA的人iPSC来源的多巴胺能神经元中产生DNA高甲基化,在体外研究中证实了其可用于治疗帕金森病[47]。而将甲基胞嘧啶双加氧酶1(Ten-eleven translocation 1,TET1)催化结构域与dCas9融合,可快速实现启动子的去甲基化,并诱导靶基因的表达上调,从而驱动各种类型的细胞中靶基因的转录激活[48]。dCas9-TET的融合蛋白被用于治疗癌症[49]并减轻体内肾纤维化[50]。这些DNA甲基化调控都是持久、可逆和可遗传的。
除了DNA的甲基化修饰可以实现基因的沉默和激活,组蛋白的修饰也可以实现类似的基因调控效果。将p300催化结构域与dCas9组成融合蛋白,该蛋白质在靶向增强子和启动子区域时能实现组蛋白瞬时和高效的乙酰化,进而增强基因的表达[51]。当dCas9与组蛋白去乙酰化酶1(Histone deacetylase 1,HDAC1)融合时,可以去除乙酰化,将其靶向KRAS可以抑制癌症的生长[52]。
最近,一种全新的基于CRISPR的表观遗传编辑技术CRISPRoff被报道[53]。研究者将KRAB和D3A-D3L分别融合于dCas9的N-/C-末端,构建出新的表观遗传编辑器CRISPRoff-V1和CRISPRoff-V2,发现CRISPRoff特别是CRISPRoff-V2能持久抑制GFP(Green fluorescent protein)报告基因的表达:CRISPRoff-V2短时表达对基因转录的抑制效果至少可持续50 d。随后研究者还设计出了CRISPRon,其有效逆转CRISPRoff介导的DNA甲基化修饰和转录抑制。CRISPRoff/on系统为控制基因表达、靶向增强子和探索表观遗传的原理提供了一个强有力的工具。
3.3 碱基编辑器
在哺乳动物细胞中,CRISPR-Cas系统通过精确剪切目标基因形成DSB,随后依靠细胞内的DNA修复系统修复双链断裂,从而实现DNA序列的改变。DNA修复采用2种相互竞争的机制即HR和NHEJ[26]。借助同源修复模板,HDR可以实现精确可控的编辑,但是它的效率相较于NHEJ修复更低;NHEJ修复不依赖修复模板,直接将2个DNA末端拼接,但在拼接的过程中会产生碱基的缺失突变(indel),无法实现基因的精确编辑,大规模的DSBs也很容易引发副作用。也就是说,DSB引发的DNA修复很难实现高效稳定的单碱基突变[54]。然而,许多致病性遗传变异是由单核苷酸变异(single-nucleotide variants, SNVs)引起的[55]。因此,仅仅通过简单的基因敲除与插入不能实现这些疾病的有效治疗,需要开发能够高效纠正SNV的方法和工具,这也要求更加精密的基因编辑技术[13, 56]。因此,先后开发出了基于CRISPR-Cas9的碱基编辑器和先导编辑器。这些工具不再依赖于DSB造成的DNA剪切,显著降低了错误修复造成的毒性,这使得CRISPR-Cas系统得到了更广泛的应用[57, 58]。
碱基编辑器(base editor,BE)是一类在单碱基水平实现特定碱基类型高效精准编辑的工具。BE能够以可编程的方式实现DNA碱基的转换,不需要切割DNA产生DSBs或同源供体模板。目前,广泛使用的DNA碱基编辑器主要是将可编程的DNA结合蛋白质与碱基脱氨酶整合在一起组成融合蛋白质。主要有2种类型:胞嘧啶碱基编辑器(cytosine base editor,CBE)和腺嘌呤碱基编辑器(adenine base editor,ABE),分别可以实现C-to-T和A-to-G此类碱基之间的转变[59]。
CBE的核心元件是由dCas9 或 nCas9蛋白(Cas9 Nickase,单链切割活性的Cas9蛋白)与胞嘧啶脱氨酶组成的融合蛋白质。融合蛋白质在sgRNA引导下到达靶序列部位,与sgRNA非配对的ssDNA结合,将该ssDNA上一定范围内(Cas9原间隔序列5个碱基范围内)的胞嘧啶脱氨变成尿嘧啶,进而通过DNA复制或修复将尿嘧啶转变为胸腺嘧啶,最终实现C-G碱基对于T-A碱基对的直接替换[59]。在肌萎缩性脊髓侧索硬化症小鼠模型中,SpCas9-CBE被用来在SOD1(super oxide dismutase 1,SOD1)中创建一个过早的终止密码子,从而减少肌肉萎缩,改善神经肌肉功能[60]。
与CBE相似,ABE的核心元件是nCas与人工定向进化的腺嘌呤脱氨酶组成的融合蛋白质,在sgRNA的引导下靶向目标DNA序列,在Cas9与靶序列结合形成R-环之后,腺嘌呤脱氨酶与跟sgRNA非配对的ssDNA结合,促使一定范围内的腺嘌呤脱氨变成肌苷,肌苷以鸟嘌呤的形式进行读码与复制,最终实现A-T碱基对于G-C碱基对的替换[61]。将ABE递送到镰状细胞病患者的造血干细胞和祖细胞(HSPCs)中,可以将SCD(sickle cell disease,SCD)的致病基因HBBS(β-globin gene,HBB)转化为非致病的HBBG(makassar β-globin),这种转变是持久的,可以最大限度减少DSB的不良后果[62]。ABE也可以用来纠正Hutchinson-Gilford早衰综合征(HGPS),将LMNA(lamin A/C gene)中的致病性突变逆转,根本性的治疗了HGPS,并且防止了外膜纤维化[63]。这些发现证明了ABE在遗传学疾病治疗上的无限潜力。
3.4 先导编辑器
CBE和ABE组合能够实现的碱基编辑仍然是有限的,为了实现12种碱基的全互换,David Liu团队开发出了能实现全碱基转化与多碱基精确缺失突变的先导编辑器(prime editor, PE)[64]。PE在CRISPR-Cas9的基础上:(1)在sgRNA的3′末端增加一段逆转录酶的带有基因编辑序列的RNA引物,组成pegRNA(the engineered guide RNA),pegRNA中包含单导向RNAs(single guide RNAs,sgRNAs),在其3′-端还有一段引物结合序列(primer binding site,PBS)和转录模板序列(RT template);(2)将dCas9与逆转录酶融合,dCas9在pegRNA上的sgRNA序列指引下,切割DNA单链,pegRNA 3′-端的PBS(引物结合序列)可以与切割断点前的互补序列识别配对,逆转录酶以pegRNA上PBS序列后的模板序列为模板进行逆转录,将目标序列直接聚合到切口的DNA链上[61]。实现任意小片段序列的插入、敲除和替换,且显著地降低脱靶率。PE的强大编辑功能使其具有巨大的治疗潜力。在α1-抗胰蛋白酶缺乏(Alpha-1 antitrypsin deficiency,AATD)的小鼠模型中,PE通过创建A-to-G编辑来去除SERPINA1(serpin family A member 1)中的致病性E342K突变[65]。PE还可以通过创建G-to-T编辑来纠正小鼠视网膜中的Dnmt1(DNA methyltransferase 1,Dnmt1)突变,这些都是其他编辑器无法替代的。
尽管BE和PE相对于传统的CRISPR-Cas方法能够脱离DSB提供更加精确的基因编辑,但是它们仍然在一定程度上诱导有害的转录反应,其中CBE的毒性最强[66]。因此,在推进碱基编辑器和先导编辑器的临床应用上,应对其安全性展开进一步评估。
3.5 靶向RNA编辑的CRISPR-Cas系统
靶向DNA的基因编辑由于其变化是永久性的,一旦发生脱靶编辑会造成不可逆转的巨大伤害,而RNA靶向的CRISPR-Cas系统(RCas)则不存在这类问题。因此,越来越多研究者开始投入到RCas系统的发掘与改造,从而推动了RNA靶向研究的快速发展[67]。目前,RCas系统大致可以分为两大类,一类是对传统DNA靶向的CRISPR系统改造,实现RNA靶向调控;另一大类则是特异性靶向识别ssRNA的CRISPR系统[68]。最具代表性的分别是CRISPR二类中的Cas9系统和Cas13系统。
3.5.1 Cas9系统 传统的CRISPR-Cas9系统依赖于sgRNA和非sgRNA互补链上的-NGG PAM位点,使其实现Cas9对DNA靶序列的特异性识别和切割[69, 70]。Cas9的这一特性限制了其对单链RNA序列的靶向编辑,加入带有PAM序列的互补反式DNA寡核苷酸(PAMmer),可以克服这一限制,能够促使Cas9与RNA高亲和力结合并切割RNA;Cas9切割效率受PAMmer长度的影响;这一策略使得CRISPR-Cas9系统实现RNA靶向序列靶向识别和切割[71]。进一步研究发现,PAMmer只影响核酸酶的RNA切割活性而不影响其与RNA的结合能力,受此启发构建的dCas9蛋白,(称作RCas9)使其在sgRNA的引导下实现RNA的靶向识别和结合,这一作用无需PAMmer参与,而且RCas9可以在无PAMmer的情况下,替换与微卫星重复异常扩增序列结合的RNA结合蛋白质,促使微卫星重复异常扩增序列降解[72, 73]。最近,有报道称CjCas9(空肠弯曲杆菌Cas9)[74]、NmeCas9(脑膜炎奈瑟菌Cas9)[75]和SauCas9(金黄色葡萄球菌Cas9)[67]可以不依赖PAM,实现ssRNA靶向,并且CjCas9和NmeCas9的ssRNA切割是可编程的。不过对于这些新来源的Cas9系统,其具体机制仍有待进一步研究。
3.5.2 Cas13系统 与Cas9需要依赖PAM的靶向切割机制不同,Cas13系统能特异性地识别并靶向ssRNA,而且Cas13-crRNA复合物在识别靶序列后,除了切割crRNA的ssRNA,Cas13还会并行切割与crRNA配对结合ssRNA附近的其他ssRNA[76, 77],因此,其在基因敲低中的应用受限。但失去酶活性的Cas13也可以与dCas9一样被用于RNA的编辑。
3.5.3 其他RNA编辑技术 SNAP-tag(DNA repair enzyme alkylguaninetransferase,AGT or SNAP-tag)与ADAR(adenosine deaminase acting on RNA,ADAR)或者APOBEC1(apolipoprotein B MRNA editing enzyme catalytic subunit 1,APOBEC1)等碱基编辑酶的融合蛋白质,在SNAP-tag的催化下可与O-6-苄基鸟嘌呤修饰的gRNA特异性结合,在RNA特定位点进行碱基编辑[78, 79]。通过gRNA上的MS2适配体招募MCP和ADAR2或者APOBEC1等碱基编辑酶形成的融合蛋白质[80],也可用于RNA特定位点的碱基编辑。
4 CRISPR-Cas递送策略
CRISPR系统能在任何遗传环境下设计一个编辑可控的gRNA引导到目标序列进行基因编辑,展现了遗传疾病治疗的强大潜力。为了实现基于CRISPR系统的体内基因编辑治疗,必须寻找到一种递送载体将它们安全、高效和准确地递送到人体相关器官和组织,并且尽可能避免触发机体的免疫反应,这是目前体内基因编辑应用的一个重大挑战[81]。
4.1 基于腺相关病毒和脂质纳米颗粒(LNPs)的基因编辑工具递送
迄今为止,已有不同递送载体用于CRISPR-Cas系统的传递,主要可以分为病毒载体(腺相关病毒、慢病毒)和非病毒载体两大类,非病毒载体包括天然合成材料(脂质体、金纳米颗粒、脂质纳米颗粒)和物理方法(电穿孔、超声穿孔法和显微注射法)2种。本文主要介绍腺相关病毒和脂质纳米颗粒(lipid nanoparticles,LNPs)。
腺相关病毒(adeno-associated virus,AAV)是通过在反向末端重复序列之间装载目标基因用于疾病治疗,这种方法会受AAV大小的限制,只允许装载大小4.7 kb以内的Cas和基因[82-84]。为了克服AAV装载大小的限制,目前有2种应对措施,一种方法是设计出更小的Cas蛋白。目前报道的CRISPR-Cas12f系统中enAsCas12f蛋白大小是SpCas9的三分之一,并且表现出比其亲本蛋白质更好的活性和低脱靶率,很大程度上克服了递送载体载荷量的限制[85, 86]。另一种则是将基因编辑组件放置到2个AAV系统中同时递送到同一个细胞,再在细胞内组合发挥编辑活性。但是这种方法的效果要明显逊色于单个系统递送[87]。除此之外,AAV在临床中的应用受其自身免疫原性影响,单次注射后会产生血清型特异性中和抗体,需保证一次足量给药就能达到预期治疗效果,这可能带来意外的不良反应[88]。2022年10月,一位27岁杜氏肌营养不良症(duchenne muscular dystrophy,DMD)患者在接受重组腺相关病毒9型(rAAV9)载体递送的CRISPR基因编辑治疗后不幸去世,高剂量rAAV引起的强烈先天性免疫反应导致了急性呼吸窘迫综合征(acute respiratory distress syndrome ,ARDS),这也是第一例CRISPR介入治疗死亡的病例[89]。
以AAV为代表的病毒载体的不足推动了LNPs在内的非病毒传递系统的发展。与AAV不同,LNP毒性和免疫原性更低并且不受载荷量的限制。它由可电离阳离子磷脂、辅助磷脂、胆固醇和聚乙二醇化磷脂四个部分组成。为了将其包裹的核酸物质运送到靶细胞,LNP首先通过内吞作用进入细胞,在内体酸化后通过破坏内体膜分泌出来,对其结构进行改造可以调控递送[90-93]。LNP存在较强的肝内自然累积效应,目前,已被美国FDA批准用于通过静脉注射向人肝细胞输送治疗性siRNA[94-96]。而其非肝靶向递送仍然局限于通过局部注射实现,设计开发非肝靶向的LNP递送系统仍是一大挑战。此外有研究表明,LNP在小鼠模型中被证明会诱导炎症恶化,并且这种恶化是时间和剂量依赖性的[90, 97][110-111],LNP递送系统的安全性需要进一步考量。
AAV的递送受限于装载量和免疫原性,LNP虽然克服了AAV的装载限制且有较低的免疫原性,但是其研究仍然不够深入,再加上CRISPR基因编辑器的长时间滞留会导致脱靶编辑造成不可逆转的伤害。因此,递送仍然是基因编辑体细胞治疗的最大瓶颈。如果在这些问题的优化方面取得突破性进展,将为基因编辑的临床转化注入新的活力[98, 99]。
4.2 细胞外囊泡与基因编辑工具递送
细胞外囊泡(extracellular vesicles,EV)是由细胞自发分泌的内源性膜性颗粒,通常作为信使在细胞之间运输各种物质,例如核酸、蛋白质、小分子代谢物和脂质等[100]。根据EV生成途径的不同,EV可以分为外泌体(exosome,30~150 nm)、微囊泡(microvesicle,100 nm-1 μm)和凋亡小体(apoptatic body, 100 nm~5 μm),其中30~200 nm的小细胞外囊泡常用于递送包括CRISPR-Cas在内的不同物质[101-103]。与传统递送载体存在自身免疫原性、毒性和易被机体代谢清除等问题不一样,EV表面所携带的蛋白质和脂质可以防止其被机体的网状内皮系统(reticuloendothelial system,RES)和单核吞噬系统(monophagocytic system,MPS)清除,从而延长半衰期。此外,EV的小尺寸使它们更容易穿过细胞外基质等物理屏障[104]。目前,利用EV包载药物用于治疗的疾病涉及癌症[105, 106]、心血管疾病[107]和神经系统疾病[108]等。与LNPs引发的炎症反应相比,EV诱导的促炎细胞因子的数量和效应都较弱,安全性更高[109]。基于以上特点,EV具有作为递送CRISPR-Cas系统的优良载体的潜力。
CRISPR-Cas系统可以通过质粒DNA、mRNA或核糖核蛋白复合物(RNP)3种不同形式进行递送,各有优缺点[110]。相较于前两者,RNP递送不需要进行转录和翻译,能直接进入细胞进行编辑,活性更高,有更低的脱靶率和免疫原性,成为目前最具潜力的递送形式[111]。研究表明,单纯的Cas9蛋白直径在10~15 nm之间,加入gRNA与之混合后会发生聚集直径达到200 nm,加入聚谷氨酸或ssODNenh后可以阻止聚集使其直径降到100 nm左右[112],这超过了大部分病毒和非病毒载体的装载能力。基于聚合物的纳米颗粒和脂质体又易在体循环中被降解,这些因素都限制了RNP的递送[113]。与病毒不同,EV可以在大小上进行修饰。混合EV已被开发用于携带CRISPR/Cas系统等大型分子。混合EV不仅货物包载能力更强,而且由于脂质体带正电荷,带负电荷的RNA和DNA能更有效地被包载到EV中[114]。EV的低免疫原性和可操作性为RNP的递送创造了可靠条件[115][129]。目前,主要通过(1)细胞内EV生成途径将CRISPR-Cas编辑系统包载到EVs;这些包载方式不会破坏EV膜结构,保证其递送性能稳定;(2)利用电转和化学转染等物理和化学方法将Cas蛋白与sgRNA形成的核糖核蛋白质颗粒(ribonucleoprotein particles,RNPs),或者能够编码表达Cas蛋白和sgRNA的质粒包载到EV中[116, 117](Fig.5),这些方法各有优势和缺点。

Fig.5 The strategies for extracellular vesicle encapsulating gene editors The CRISPR-Cas system can be delivered in three different forms: plasmid DNA, mRNA, or ribonucleoprotein complex (RNP), and can be loaded into the EV in two ways. Encapsulation of the Cas9 RNP by physical/chemical approaches: Membrane disruption of the EV was first performed and then the CRISPR-Cas editor was loaded through a concentration gradient way; encapsulation of the CRISPR-Cas editing system through EV biogenesis (this approach does not damage the EV membrane structure and ensures stable delivery performance)
基因编辑工具的精准靶向递送是精准、高效和靶向基因编辑的难点。EV具有归巢靶向其亲本细胞同源组织的特性,利用这一特性,可以采用EV实现组织和器官的靶向递送。例如,来自小胶质细胞的EV倾向于到达中枢神经系统[118],来自施万细胞的EV倾向于靶向周围神经[119],而来自肿瘤细胞的EV倾向于到达同源的肿瘤细胞[120]。这种靶向倾向可能与EV同母细胞共享表面受体和基质结合蛋白质有关,因此,根据递送目的选择合适来源的EV是递送设计的重要考虑因素。除此之外,采用靶向蛋白质或靶向肽对EV进行表面修饰也能实现EV组织或器官的靶向递送。在EV表面表达具有靶向性的重组蛋白质,例如将EV膜蛋白Lamp2b(lysosome-associated membrane glycoprotein 2b)、PDGFR(platelet-derived growth factor receptors)、CD63(CD63 antigen)与受体细胞表面的配体融合可以实现EV对特定受体细胞的靶向递送。Mentkowski等人将一组心肌细胞靶向多肽与Lamp2b融合,所得的EV经给药后可提高其心脏靶向递送效率,EV在心脏的保留率提高2.4倍[121]。Liang等人将软骨细胞亲和肽DWRVIIPPRPSA和Lamp2b融合的重组蛋白组表达到EV上,赋予了其靶向软骨组织治疗骨关节炎的能力[122]。利用人PDGFR的跨膜结构域作为载体与2种靶向CD3和EGFR(epidermal growth factor receptor)的scFv抗体串联融合,通过EV生成途径表达到EV膜表面,可以实现乳腺癌的靶向治疗[123-125]。除此之外,整合素的表达也与组织器官的靶向递送相关,特定部位的整合素表达下降会减少该组织对EV的摄取。例如,ITGβ4是一种EV上表达的肺相关整合素,Hoshino等人发现ITGβ4(Integrin subunit beta 4)的下调显著降低了体内肺组织对EV的摄取;相反,过表达ITGβ4的骨转移细胞系会促进EV进入肺[126]。
尽管与AAVs或LNPs载体相比,EV在体内生物相容性和稳定性方面具有许多优势,但基于EV的CRISPR递送仍处于临床前阶段,仍然存在几个局限性需要克服。目前与EV的生物学功能与作用机制仍有待阐明,EV生产加工优化的方法研究仍需更多研究[127]。EV具有高度异质性,不同来源的EV的生物效应可能不同,但目前缺乏通用的样本收集方案和生物标志物用以区分和表征不同来源的EV[128]。此外,EV的货物包载、靶向、摄取和释放等方面的机制仍有待进一步深入研究[129]。未来应当深入研究EV的靶向递送机制,寻找更加高效的EV货物包载方法,在实现精准递送的同时增加其摄取和释放效率。
5 基因编辑技术在糖生物学中的应用
糖类物质与蛋白质、核酸和脂质是构成生命的四大基本物质。与DNA和蛋白质不同,糖链的生物合成是非模板依赖的,主要依赖级联的酶促催化合成。此外,糖链的链接方式多样,单糖种类较多,这使得糖类物质结构远比核酸和蛋白质复杂,这一特性也严重阻碍了糖类物质结构与功能的研究。研究发现,糖类物质生物合成酶基因表达水平的改变跟糖链结构的改变通常一致,但不完全相同[130, 131]。传统的RNAi基因沉默策略通常只降低转录和蛋白质水平,即使酶蛋白减少80%~90%也可能不会对糖基化产生明确和明显的影响[132]。因此,可以采用基因编辑技术对糖基化途径进行精准编辑,促进对糖类物质生物合成、结构与功能的研究,也有助于转化糖科学的发展。
5.1 糖基化修饰及其作用
哺乳动物中至少有一半的蛋白质被糖基化,各种单糖残基在糖基转移酶和糖苷酶的催化下连接到蛋白质、脂质或核酸上的过程称为糖基化(glycosylation)。在糖基转移酶(glycosyltransferase, GT)的参与下,真核生物细胞中的绝大多数蛋白质糖基化都沿着分泌途径发生,从内质网开始,在高尔基体中完成。大多数GTs是II型跨膜糖蛋白质,兼具内质网和高尔基腔导向的催化结构域,利用活化的糖核苷酸作为供体,以囊泡形式从内质网运输至高尔基体,参与糖基化修饰[133-135]。N-糖基化是真核生物中一种普遍的糖基化形式。N-糖基化在内质网中由与STT3A或STT3B催化亚基组装的寡糖转移酶(oligosaccharyltransferase,OST)复合物启动,该复合物将寡糖转移到多肽主链上,在含有N-X-S/T序列的Asn残基上被修饰,再转移到高尔基体中以多种方式进行进一步修饰[136-138]。在真核生物中,除了O-GalNAc和O-Xyl (proteoglycan)糖基化在高尔基体进行外,大多数类型的O-糖基化也都是在内质网中由不同的多肽糖基转移酶启动的,随后在高尔基体中进一步进行延伸、分叉和终止[133, 139]。这些过程可被小分子糖类似物抑制[140-142](Fig.6)。

Fig.6 Cellular glycoengineering Glycosylation can be modulated by direct editing of the GT gene using precision gene editing techniques. Some artificial sugar analogues can modulate the process of glycosylation metabolism (core extension, elongation and branching of oligosaccharides). Extracellularly, glycans may be remodeled by selective endo-/exo-glycosidases. Mutant cells with loss/gain of different glycosylation profiles can be derived using cytotoxic lectins
糖基化异常已被发现与癌症等众多疾病的发生发展密切相关[143-147]。蛋白质糖基化异常会导致蛋白质折叠错误、稳定性降低和功能丧失[148]。人类基因组包含大约700个基因参与细胞糖基化相关酶和转运体等功能蛋白质的编码[149],其中超过200个基因编码GTs[150]。这些基因功能的异常还可导致先天性糖基化异常疾病(congenital disorders of glycosylation,CDG),CDG已发现接近200种,多数是罕见病,涉及不同的N-糖基化和O-糖基化(O-mannose、O-glucose、O-fucose、O-GlcNAc、O-GalNAc、糖胺聚糖、糖基磷脂酰肌醇和糖脂)过程[151],这些疾病多数缺乏有效治疗手段[151, 152]。精确基因编辑技术的发展为细胞糖基化的研究和相关疾病的治疗提供了无限机会。
5.2 基因编辑技术在糖生物学中的应用
基因编辑在糖生物学研究中的应用具有悠久历史[132]。早期糖基转移酶基因或其他影响糖基化的基因编辑是通过随机突变和凝集素抗性筛选等随机筛选技术实现[153-155]。随着基因编辑技术的发展,与其他领域一样,糖生物学领域的基因编辑也经历了基于胚胎干细胞中的同源重组策略,对糖基化相关基因进行靶向敲入或敲除(knock out, KO / knock in, KI)[143, 156]到基于可编程核酸酶基因编辑的转变,逐步实现了高效、精准和靶向编辑。这些技术极大地促进了糖类物质生物合成、结构与功能研究和糖复合物的生产和应用。
5.2.1 糖类物质功能、生物合成及机制解析 N-乙酰半乳糖胺(GalNAc)型O-糖基化是最复杂的蛋白质糖基化形式之一,有多达20种不同的GalNAc-T亚型控制O-糖基化位点,目前的分析方法均不能很好地理解和区分不同GalNAc-T家族成员的功能及作用机制。精确基因编辑技术的发展为研究同工酶的功能和机制提供了手段。Schjoldager等人[157]通过敲除HepG2(human hepatocellular carcinomas)中GALNT1(polypeptide N-Acetylgalactosaminyltransferase 1)和GALNT2(polypeptide N-acetylgalactosaminyltransferase 2)来确定该细胞中ApoC Ⅲ(apolipoprotein C-III)是GALNT2的底物。近来,有研究利用SimpleCell策略,构建了只表达Tn-O-糖型的癌症细胞系,并以该细胞作为抗原制备糖型确定细胞的特异性单克隆抗体,这为癌症免疫疗法提供了新策略[169]。此外,CRISPR-Cas9基因编辑技术的出现可以实现全基因组功能筛选,不仅能鉴定不同的糖基转移酶基因,还可以发现新的糖基化基因和功能[158]。Han等人通过人肺上皮细胞基因组规模的CRISPR/Cas9敲除筛选,发现了参与唾液酸生物合成和相关糖基化途径的调控基因,包括SLC35A1(solute carrier family 35 ember A1)[159]。Kelkar等人开发了一种混合基因筛选系统发现MGAT5(alpha-1,6-Mannosylglycoprotein 6-Beta-N-acetylglucosaminyltransferase)产生的四天线N-聚糖,参与CD8+ T细胞对胶质母细胞瘤肿瘤细胞的杀伤[160]。基因组筛选也发现了硫酸乙酰肝素生物合成的新调控因子KDM2B(lysine demethylase 2B)[161]和ZNF263[162]。
5.2.2 应用于糖蛋白质组学分析的SimpleCell策略 蛋白质糖基化分析的最大难点是糖肽的富集,而聚糖的多样性和异质性阻碍了富集的进行。目前,已有的富集方法包括凝集素层析[163]和基于酰肼法[164]与硼酸法[165]的化学富集等方法。随着基因编辑技术的发展,可以通过理性糖基化工程从源头上简化或消除糖链结构的多样性和异质性,从而实现糖肽的有效富集。这种简化细胞糖链的策略被命名为SimpleCell策略,即利用基因编辑技术,生产能够合成糖型确定的糖蛋白质组的工程化细胞。该策略通过凝集素层析从细胞裂解液和分泌组中富集糖肽,并利用高能碰撞诱导解离或ETD模式,通过LC-MS/MS快速灵敏地鉴定糖基化位点[132]。
SimpleCell策略最初被用于N-乙酰半乳糖胺型O-糖基化,通过敲除COSMC(T-synthase-specific molecular chaperone)基因实现去O-糖基化,进而使O-聚糖被截断成易被凝集素有效捕获的结构,该结构也可用于糖基化位点的鉴定[166]。随后,SimpleCell策略通过敲除POMGNT1(protein O-linked mannose N-Acetylglucosaminyltransferase 1)实现O-甘露糖基化糖肽的鉴定,将复杂的O-甘露糖基化糖链简化为只含甘露糖残基的糖肽[167],简化后的结构能被ConA凝集素富集,由此发现,钙粘蛋白和丛状蛋白超家族蛋白质被O-甘露糖基化。这些O-甘露糖基化在囊胚形成过程中对E-钙黏着蛋白介导的细胞粘附至关重要[168]。Konstantinidi等人利用基因编辑技术敲除COSMC或C1GALT1(core 1 synthase, Glycoprotein-N-Acetylgalactosamine 3-Beta-Galactosyltransferase 1)基因获得HEK293 SimpleCells,用于确定粘蛋白质和粘蛋白样糖蛋白的O-糖结构域,结果发现,粘蛋白串联重复序列内所有潜在糖基化位点上均能被有效地O-糖基化[170]。
然而SimpleCell策略在广泛应用的同时也存在一些问题。借助基因编辑技术设计出简化糖结构的SimpleCells,在获取糖基化位点信息的同时丢失了糖链的结构信息[171],并且细胞中糖基化途径的遗传截断可能会影响早期的生物合成步骤[172]。对于前者,非数据依赖采集模式的质谱分析有望解决这个问题[173],而对于后者,目前暂未发现SimpleCells和野生型细胞在O-糖蛋白质糖基化位点的显著差异。总之,SimpleCell策略的广泛应用大力地推动了糖蛋白组学的发展。
5.2.3 细胞糖芯片的构建和应用 细胞表面糖类物质在细胞-细胞和细胞-基质的相互作用方面具有重要作用,并对免疫细胞的迁移、胚胎发育和癌症转移等过程至关重要。糖类物质通常通过与蛋白质相互作用发挥作用。糖结合蛋白质 ( glycan-binding proteins, GBPs ) 主要分为凝集素和糖胺聚糖结合蛋白质。
糖芯片是一种研究GBPs结合特异性以及蛋白质与糖类物质相互作用的技术[174-176],传统糖芯片通常是通过化学偶联将糖类物质固定在特定的基质材料上而得,类似于DNA芯片[177-179]。与糖芯片结合的蛋白质通常使用生物素化或荧光标记的抗体检测。因而可以用于筛选发现糖结合蛋白质,具有样品用量少和高通量的特点。缺点是传统糖芯片不能模拟糖复合物和细胞表面的自然环境,细胞糖芯片弥补了这一不足[180]。通过GT基因的KO/KI获取具有不同糖基化特征的等基因细胞文库,构建细胞糖芯片。细胞表面聚糖密度、空间排列、与脂质或蛋白质的附着,以及相互竞争的聚糖结构的存在均可显著影响聚糖的识别和结合能力[181],细胞糖芯片因而可以更好地再现糖链在自然条件下的作用状态,因此,作为传统糖芯片的互补方法得到广泛应用。
采用一个人源糖基转移酶基因的CRISPR/Cas9 gRNA库[182],Narimatsu等人[183]基于CHO细胞中开发了一个糖胺聚糖细胞文库,命名为GAGOme。该文库可用作糖胺聚糖细胞糖芯片、用于重组表达含不同糖胺聚糖(GAG)结构的蛋白质聚糖,以及表达不同结构的GAG链用于制备传统的糖胺聚糖芯片。该团队[180, 182]构建了一个糖基工程化改造的等基因HEK293细胞文库用作细胞糖芯片,利用该细胞文库发现,微生物黏附素与O-聚糖簇斑块在真实细胞环境下的结合特征。在这些研究的基础上,哥本哈根大学与江南大学2个课题组先后[142, 184]利用基因编辑技术构建了HEK293中的N-和O-糖基化图谱,将173个人类GTs分为16个不同的途径和主要生物合成步骤,该图谱将细胞中GTs的表达数据从单细胞转录组学转化为可预测的糖基化能力。
硫酸乙酰肝素(heparan sulfate,HS)的结构功能关系研究极具挑战,作者团队[185]通过单独或组合敲除表达HS的基因,建立了一个能够表达不同硫酸乙酰肝素结构的小鼠肺内皮细胞文库,共包含18个细胞系。利用该文库,作者团队进一步阐明了HS对FGF2-FGFR1(fibroblast growth factor 2-fibroblast growth factor receptor 1)信号转导的调控作用及分子机制,同时也确定了抗HS单链片段可变区抗体(single chain fragment variant, scFv)的表位特征。借助该文库,作者团队[186]还确定了与Furin结合的HS结构特征,进一步解释了Furin的工作机制,为基于Furin的药物发现提供了新方向。研究显示,SARS-CoV2型病毒刺突蛋白通过其受体结合域(RBD)与细胞HS相互作用,Clausen等人[187]利用CRISPR-Cas9技术制备的能表达不同HS结构的Hep3B细胞文库,不仅证实新型冠状病毒的附着和侵袭具有HS依赖性,而且确定了与RBD蛋白结合的HS的结构特征[188],这提供了新的抗病毒治疗策略。
5.2.4 蛋白质糖基化工程改造与蛋白质药物生产 哺乳动物细胞糖基化改造的目的主要是减少糖基化的异质性、敲除非人源糖链生成基因和生产具有特定糖型的蛋白质[189, 190]。
CHO细胞糖基化系统与人源细胞类似,目前,大多数蛋白质药物采用中国仓鼠卵巢(Chinese hamster ovary, CHO)细胞系生产[191, 192]。CHO生产的蛋白质药物主要为IgG抗体,IgG 抗体Fc中的一个保守位点 Asn 297的N-聚糖与Fc受体的结合至关重要,是抗体功能的决定因素之一[193]。该位点N-聚糖的糖型是抗体与FcγRIIIa结合的重要因素[194]。其中平分型糖链是结合必须的,而末端的岩藻糖残基则抑制抗体与FcγRIIIa的结合,补体介导的细胞毒作用需要末端的半乳糖残基。敲除FUT8(fucosyltransferase 8)基因的CHO细胞可以产生无岩藻糖基化修饰的抗体,该抗体能够产生更强的抗体依赖细胞毒作用(antibody dependent cytotoxicity, ADCC)[190, 195, 196]。第一个实现FUT8基因敲除的CHO细胞系是通过靶向同源重组产生的,这种方法需要筛选鉴定超过10 000个CHO克隆才能获得敲除细胞[196],而基于CRISPR-Cas的精准基因编辑技术能够快速构建基因突变细胞系[197]。利用基因编辑技术分别构建FUT8和β4GALT1(beta-1,4-galactosyltransferase 1)分别敲除和双敲除的CHO细胞系,这些细胞可以用以生产不同半乳糖修饰和岩藻糖修饰的抗体[198]。利用CRISPR-Cas9技术也构建了UDP-Gal-4-epimerase(Gale)和GDP-L-fucose synthase(Fx)分别敲除和双敲除的CHO细胞系,这些细胞不能合成UDP-Gal或/和GDP-Fuc,也可以用于生产不同半乳糖修饰和岩藻糖修饰的抗体[199]。除了上述的2种N-聚糖修饰,N-聚糖的唾液酸化可能影响免疫调节功能和循环半衰期。人HEK293-T细胞能产生α2-3-和α2-6连接唾液酸的混合物,大多数可溶性糖蛋白质(包括IgG)表达α2-6连接唾液酸[190]。研究显示,具有α-2,6唾液酸化的免疫球蛋白G对抗体依赖性细胞毒性和抗炎功效有影响[200, 201]。因此,设计匀质α2-6-唾液酸化糖型的IgG抗体有助于介导抗炎作用并改善血清半衰期。然而,由于CHO细胞中仅存在α-2,3唾液酸转移酶[202],且唾液酸转移酶不能修饰重链N-聚糖,因此,难以改造生成α-2,6唾液酸化抗体。过表达α-2,6唾液酸转移酶(ST6Gal1基因编码, ST6 Beta-Galactoside Alpha-2,6-Sialyltransferase 1),同时使用CRISPR-Cas9技术敲除ST3GAL4(ST3 Beta-Galactoside Alpha-2,3-Sialyltransferase 4)和ST3GAL6(ST3 Beta-Galactoside Alpha-2,3-Sialyltransferase 6)基因,在CHO细胞系中成功生成α-2,6唾液酸化IgG抗体[203]。虽然CHO细胞确实能产生安全有效的糖蛋白质药物,但由于所得N-聚糖的异质性使得质量难以控制[204]。目前,CHO基因工程正朝着增强糖基化能力,减少异质性的方向前进,而精确基因编辑技术的出现促进了这一进程[205]。Zhang等人[206]采用基因编辑技术敲入/敲除了CHO细胞N-糖基化途径的关键的19个GTs基因,构建了相应的细胞株,这使得生产特定糖基化修饰的糖蛋白质成为可能。HEK293的糖基化工程细胞文库也可以用于特定糖型蛋白质的表达[142][184]。
6 问题与展望
CRISPR-Cas系统作为最具潜力的基因编辑工具,在遗传疾病治疗上有着显著优势,并且已经在临床前和临床治疗中得到了广泛的应用[207]。CRISPR-Cas基因组编辑也是创建用于人类遗传疾病研究和治疗动物模型的强大工具[208]。CRISPR-Cas等精确基因编辑技术也为糖生物学研究提供了新工具,为基础糖生物学研究及糖类相关疾病治疗开辟了新道路。例如,基因编辑技术为治疗用重组糖蛋白质的提质增效提供了新手段。然而,作为一种新工具,CRISPR-Cas仍然还存在许多问题亟待解决:(1)传统的基因编辑技术依赖于DSB,无法实现碱基水平的插入、删除和替换,最近几年新兴的CRISPR工具虽然克服了上述限制,但仍然无法摆脱其自身毒性。由于这些工具对于基因的调控在DNA水平,一旦出现错误编辑,其伤害是终身甚至可遗传性的。(2)CRISPR的精准靶向递送仍然是一个难点,其主要障碍包括低递送效率和缺乏组织特异性,这严重限制了其临床应用。目前,应用最多的基于AAV和LNP递送策略,其受载量、自身免疫原性、易受到达靶细胞前被循环系统清除等限制。EV凭借其体内生物相容性和稳定性方面等许多优势,在一定程度上比AAV和LNP具有优势,随着EV摄取、释放和加工等机制的进一步阐明,EV递送 CRISPR-Cas系统的应用将会得到更加广泛。(3)新的CRISPR系统仍在不断被发现,除了高编辑效率的Cas9系统、迷你型Cas12f系统和专门用于RNA编辑的Cas13系统,未来可能还有发现更多功能特异的CRISPR系统。因此,亟需对各类系统进行更加深入的研究。(4)CRISPR-Cas等精确基因编辑技术的发展为糖类物质的生物合成机制、结构与功能研究和特定糖型糖复合物的生产带来了新机会,为基础和转化糖科学研究创造了新机遇,未来需要更好地利用基因编辑技术开展合成糖生物学研究,实现格物致知向造物致知的转变,同时也开展糖基化异常疾病的基因治疗。
