不同产地滇黄精高效液相色谱法指纹图谱的建立及其多糖含量测定*
2024-04-22马琴郭思潍杜强杨国海王才武周雪吴林菁
马琴, 郭思潍, 杜强, 杨国海, 王才武, 周雪*, 吴林菁*
(1.贵州医科大学 药学院 天然药物资源优效利用重点实验室 & 贵州省高等学校天然药理与成药性评价重点实验室, 贵州 贵阳 561113; 2.贵州省农业科学院, 贵州 贵阳 550025; 3.天柱县三方田种植专业合作社, 贵州 天柱 556608)
滇黄精(PolygonatumkingianumColl. et Hemsl.)是百合科黄精属草本植物,收载于历版《中国药典》[1-2],其干燥块茎可加工成传统中药,是我国知名的“药食同源”类植物[3-4]。有研究表明滇黄精有效化学成分多样[5-6],主要含有多糖、黄酮、皂苷等化学成分[7-8],具有保护肾脏、保护心脏、抗糖尿病及抗衰老等药理作用[9-12]。滇黄精活性成分多样,且作用机制复杂,不同种属和产地的生物功能活性和成分含量也会有所差别,主要分布在云南、贵州等地[13]。目前广西、四川、云南及贵州各地区均有种植[14-16],而不同的种植生态环境是否会对滇黄精的质量产生一定影响,目前尚未见相关研究报道。中药指纹图谱是目前被国内外均认可的有效质量控制方法[13],其技术涉及众多方法,其中高效液相色谱法(high performance liquid chromatography, HPLC)具有高效迅速、重现性好及灵敏度高等多个优点,当前已成为中药指纹图谱研究中的首选方法[17]。由于野生资源逐渐无法满足市场需求,因此人工栽培滇黄精的经济效益逐渐显现,具有良好的发展前景[18-20]。滇黄精喜阴湿的气候环境,具有耐寒、怕干旱等特性,为明确滇黄精不同产地和有效成分含量的关系,本研究基于HPLC建立贵州不同产地及临近省份广西、四川的滇黄精指纹图谱,并结合主要有效成分黄精多糖的含量测定,希望能为有效推动高质量滇黄精人工种植的发展提供一定参考依据。
1 材料与方法
1.1 实验材料
1.1.1药材来源 试验样品收集于贵州各地,并经贵州民族大学民族医药学院王祥培教授鉴定为百合科植物滇黄精PolygonatumkingianumColl.et Hemsl.,样品来源详见表1。
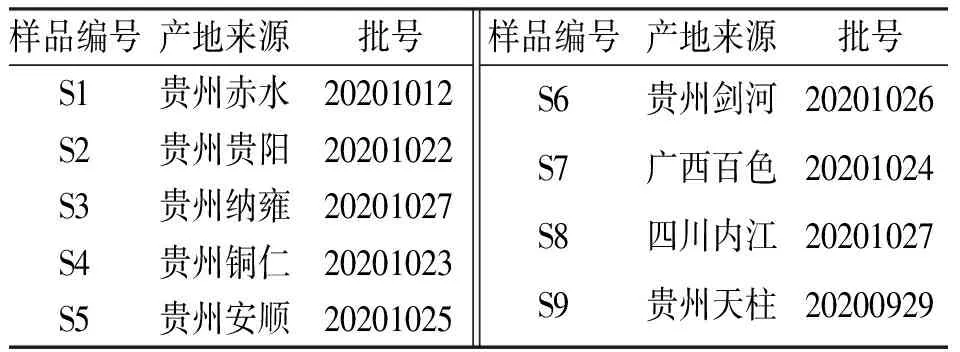
表1 不同滇黄精药材样品的产地来源及批号Tab.1 Source and batch No. information of nine batches of Polygonatum kingianum
1.1.2主要试剂与仪器 无水葡萄糖(批号CHB190213,HPLC≥98%)购自成都克洛玛生物科技有限公司,甲醇、乙腈是色谱纯,其余试剂均是分析纯,水是纯化水;UV-2700紫外可见光分光光度计[岛津仪器(苏州)有限公司],Ultimate 3000 HPLC高效液相色谱仪(美国 Thermo Fisher Scientific公司,配备系统控制器、脱气组件、自动进样器、柱温箱、温控样品室、四元梯度泵、Chromeleon色谱数据工作站),SH2-D(Ⅲ) 循环水式多用真空泵(郑州豫华仪器制造有限公司),RV3 ECO S096旋转蒸发器[德国IKA(广州)仪器设备有限公司],AE-240型电子天平[Mettler toledo国际贸易(上海)仪器有限公司],KQ5200E型超声波清洗器(昆山市超声仪器有限公司)。
1.2 研究方法
1.2.1色谱条件 色谱柱为DiKMA Platisll ODS C18(250 mm×4.6 mm,5 μm),流动相为乙腈(A)-0.05%磷酸水溶液(B),体积分数线性梯度洗脱:0~30 min(10%~22%A)、30~50 min(22%~25%A)、50~75 min(25%~38%A)、75~80 min(38%~46%A)、80~90 min(46%~84%A)、90~105 min(84%~100%A);进样量20 μL,流速1 mL/min,柱温35 ℃,检测波长203 nm。
1.2.2供试品溶液的制备 精密称取黄精药材粉末1 g,置三角烧瓶,加80%乙醇10 mL,室温超声处理30 min(功率250 W,频率40 kHz),过滤取滤液,80%乙醇40 mL溶解滤渣,超声处理30 min,合并2次滤液,60 ℃水浴蒸干,50%甲醇定容至5 mL,摇匀,0.22 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
1.2.3HPLC指纹图谱的建立 (1)精密度试验:精密吸取滇黄精供试品溶液(S9),按“1.2.1”项下色谱条件连续进样测定6次。(2)稳定性试验:精密吸取滇黄精供试品溶液(S9)于第0 h、2 h、4 h、8 h、12 h、24 h时依次进样测定。(3)重复性试验:按“1.2.2”项下方法平行制备6份S9号滇黄精供试品溶液,依次进样测定。分别对各样品共有峰保留时间和峰面积进行统计并计算相对标准偏差(relative standard deviation,RSD)。(4)对照指纹图谱的建立:取各批次滇黄精供试品溶液进样测定,记录色谱图。采用“中药色谱指纹图谱相似度评价系统(2012版)”建立9批不同产地滇黄精指纹图谱,并统计各样品的共有色谱峰及计算相似度。
1.2.4多糖对照品溶液的制备及线性关系考察 参照2020年《中国药典》方法[1]配制葡萄糖对照品溶液,方法稍加改进,精密称取无水葡萄糖10 mg于100 mL量瓶,加水溶解并稀释至刻度,摇匀,即得多糖对照品溶液。精密吸取适量对照品溶液于5 mL试管,加水至1 mL,摇匀,冰水浴中滴加0.2%蒽酮-硫酸溶液至刻度,混匀、放冷,水浴保温10 min,取出置冰水浴10 min,即得各浓度对照品溶液;于紫外-可见分光光度计582 nm波长处测定吸光值,以吸光度值为纵坐标,浓度(mg/L)为横坐标,绘制标准曲线。
1.2.5滇黄精多糖含量测定 (1)供试品溶液的制备:分别取各批次滇黄精样品0.25 g,加80%乙醇150 mL,加热回流1 h,趁热滤过,残渣用80%乙醇洗涤3次;残渣及滤纸置烧瓶中,加水30 mL,置沸水浴中加热回流1 h,趁热滤过;残渣及烧瓶用热水洗涤4次,合并滤液与洗液,放冷,转移至250 mL量瓶,加水定容,取1 mL置5 mL试管,照“1.2.4”项下自“加水至1.0 mL”起进行制备。(2)精密度试验:取滇黄精供试品溶液(S9),重复测定6次得各样品吸光度值。(3)稳定性试验:取滇黄精供试品溶液(S9),分别于第0、20、40、60、80、100及120 min测定吸光度。(4)重复性试验:制备S9号滇黄精供试品溶液6份,依次测定各样品吸光度。(5)加样回收率:精密吸取S9号滇黄精溶液9份,测定各样品吸光度并计算多糖含量;根据多糖含量的50%、100%及150%质量分数设定低、中、高3个含量梯度,加无水葡萄糖对照品,每组平行制备3份,测定吸光度,并计算多糖加样回收率。(6)滇黄精多糖含量测定:取各批次滇黄精供试品溶液,测定各样品吸光度并计算多糖含量。
1.3 统计学分析
采用 Excel 2013 软件进行数据处理;标准曲线利用Origin 2021软件进行绘图;通过SPSS 20.0软件进行 Pearson 相关分析和系统聚类分析及主成分因子分析。
2 结果
2.1 滇黄精HPLC指纹图谱的建立
2.1.1HPLC指纹图谱的构建及共有峰的确认 滇黄精HPLC对照指纹图谱图和9批次样品叠加图谱见图1,共标定了12个共有色谱峰,设定保留时间适中、分离度较好、峰面积较大及峰形较稳定的7号共有峰作为参比峰(S),计算指纹图谱中各共有峰相对保留时间(表2)和相对峰面积(表3);9批不同产地滇黄精样品相似度介于0.762~0.946,除S8号样品外,其余8批滇黄精相似度均在0.8以上(表4)。
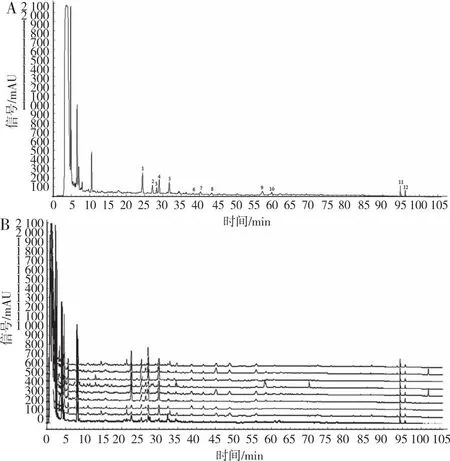
注:A为对照指纹图,B为9批次样品叠加指纹图。
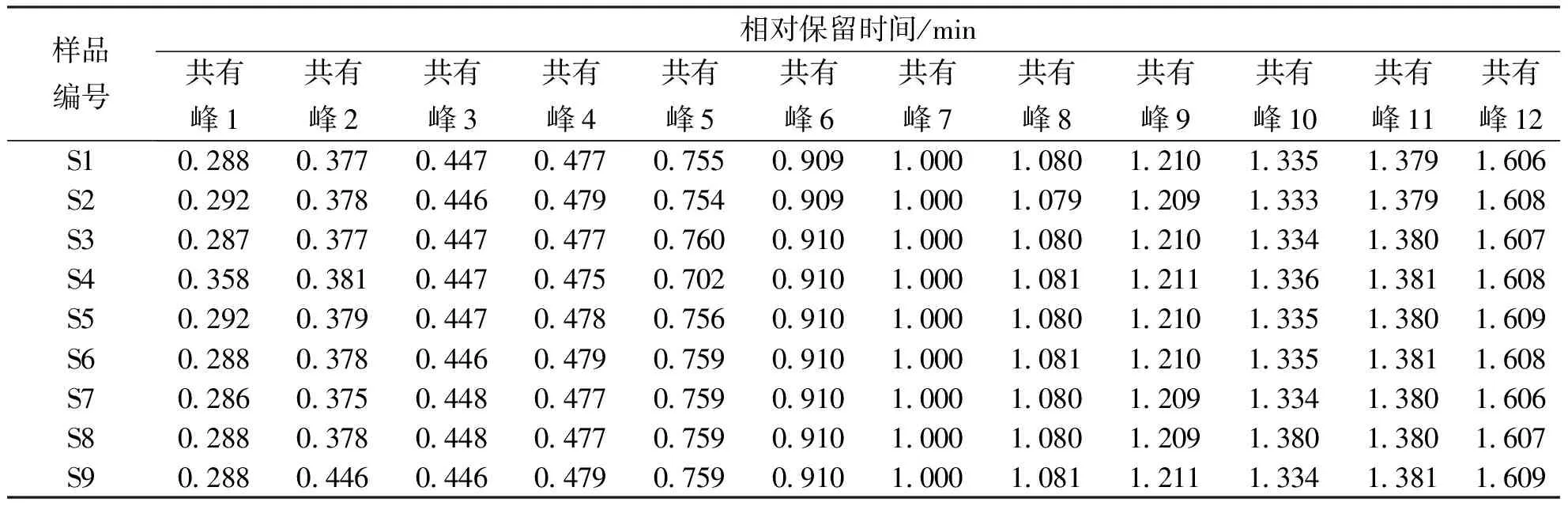
表2 不同产地滇黄精样品共有峰的相对保留时间Tab.2 Relative retention time of common peaks of Polygonatum kingianum from different origins
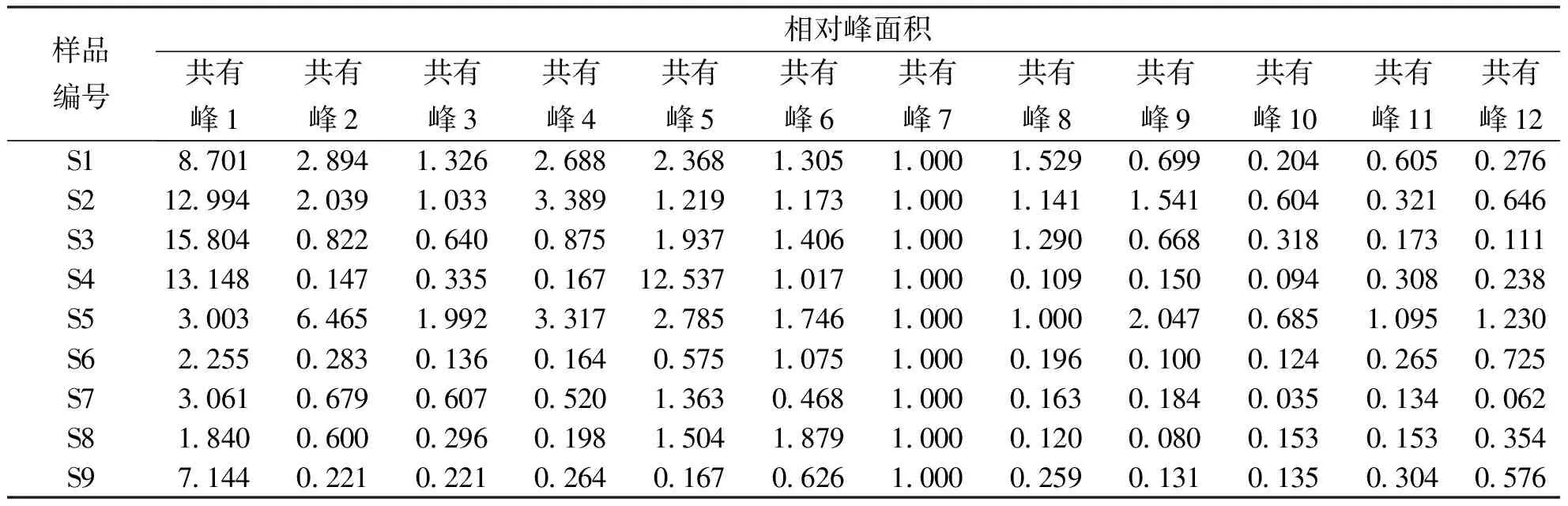
表3 不同产地滇黄精样品共有峰的相对峰面积Tab.3 Relative peak areas of common peaks of Polygonatum kingianum from different origins
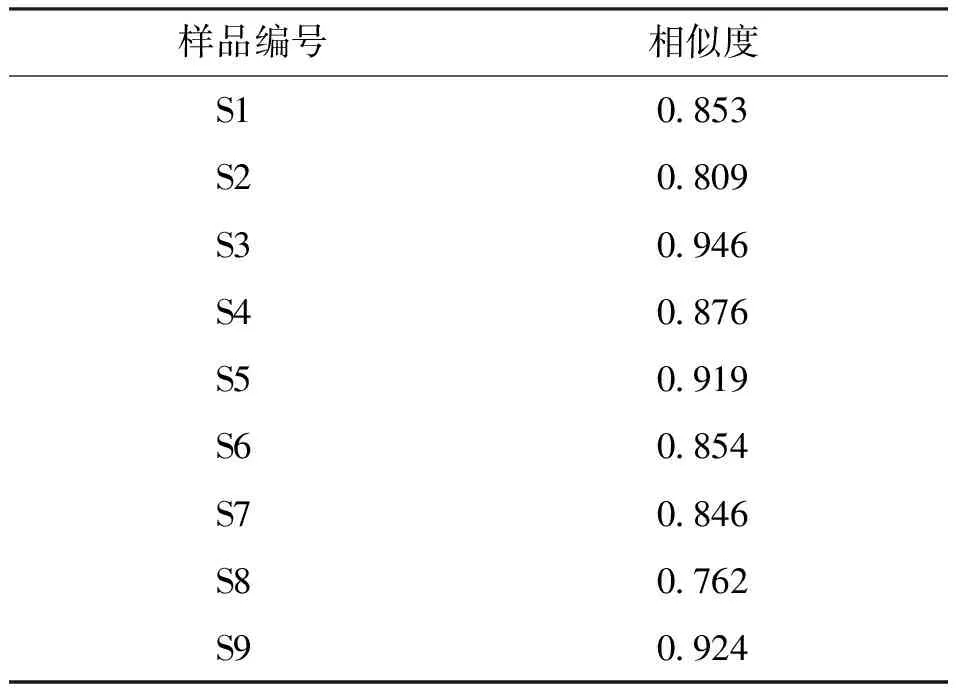
表4 不同产地滇黄精样品指纹图谱的相似度Tab.4 Similarity of fingerprint profiles of Polygonatum kingianum from different origins
2.1.2方法学验证 精密度试验测得滇黄精峰面积和共有峰保留时间的RSD值范围分别为0.30%~2.24%和0.08%~0.55%,提示本研究所用仪器平行性好、精密度高;重复性试验测得滇黄精峰面积和共有峰保留时间RSD值范围分别为2.13%~2.70%和0.02%~0.58%,提示本法的重复性良好;稳定性试验测得滇黄精峰面积和共有峰保留时间RSD值范围分别为0.80%~2.83%和0.12%~0.53%,提示供试品在24 h内稳定。
2.1.3聚类分析 滇黄精样品共有峰峰面积的系统聚类分析结果表明(图2),当欧氏距离>10时,不同来源的9批滇黄精最终分为3类,贵州贵阳、广西百色、贵州剑河、贵州铜仁及四川内江聚为一类,贵州赤水和贵州安顺聚为一类,贵州纳雍和贵州天柱聚为一类。
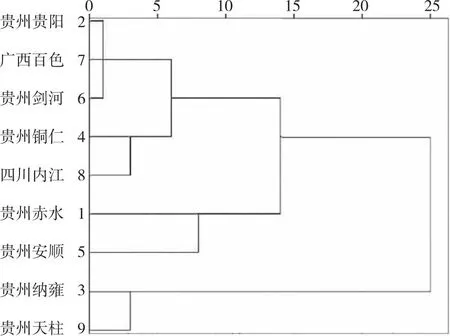
图2 不同产地滇黄精样品共有峰峰面积的聚类分析Fig.2 Cluster analysis of common peak areas of Polygonatum kingianum of different origins
2.1.4主成分分析 由表5可知,共有3个主成分因子的特征值符合要求,分别为5.273、3.069及2.118;3个主成分因子的累积方差贡献率为 87.162%,说明前3个主成分可代表样品大部分信息,适用于主成分分析;依据主因子载荷绝对值越大,对主成分贡献越大,由表6可知,主成分因子1反映了峰2、3、4、8、9、10及11的信息,主成分因子2反映了峰 6、7、11及12的信息,峰 1、5及10在主成分因子3上有较高的载荷,载荷图详见图3;通过3个主成分因子对9批滇黄精样品进行了综合评分,结果见表7,主成分因子综合得分(major factor synthesis score,MFSC)中S9号滇黄精(贵州天柱)最高,相较而言,贵州滇黄精整体质量较好,特别是贵州天柱、贵州赤水、贵州纳雍及贵州铜仁产地。
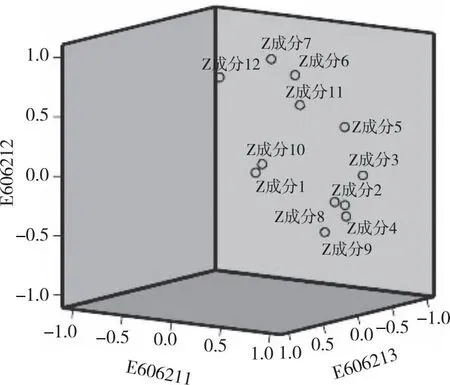
图3 滇黄精的主因子载荷Fig.3 Principal factor loadings of Polygonatum kingianum

表5 滇黄精主成分因子的特征值和方差贡献率Tab.5 Eigenvalues and variance contributions of the principal component factors of Polygonatum kingianum
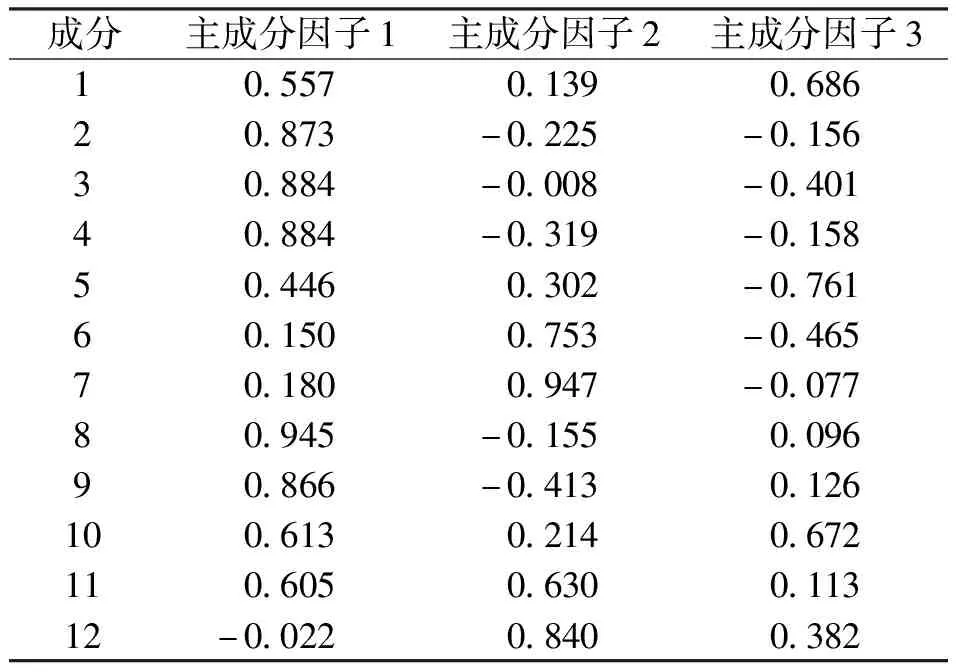
表6 旋转后的滇黄精主因子载荷矩阵表Tab.6 Rotated principal factor loading matrix for Polygonatum kingianum
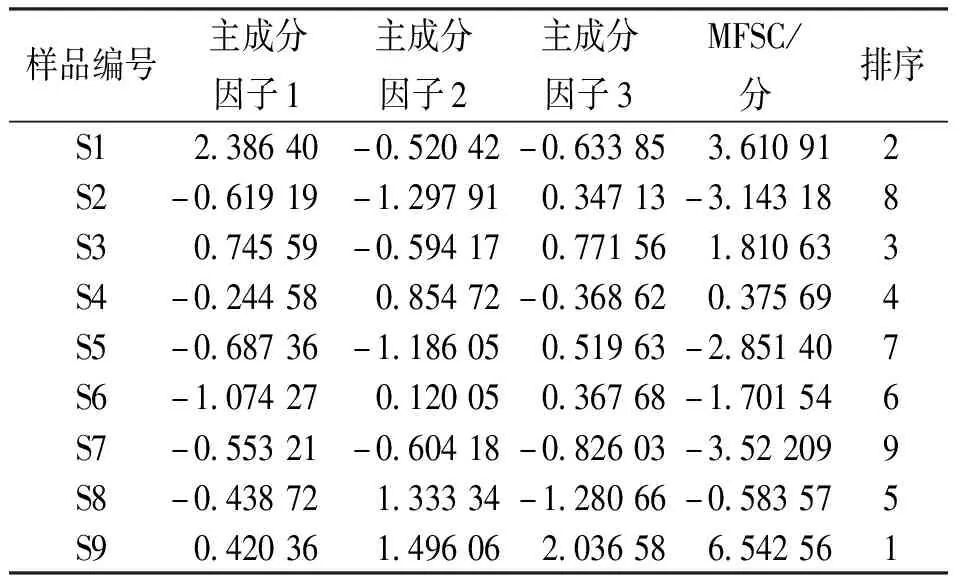
表7 滇黄精的主成分因子得分及排序Tab.7 Principal component factor scores and ranking of Polygonatum kingianum
2.2 滇黄精多糖含量测定
2.2.1多糖标准曲线的绘制 按照“1.2.4”项下方法对梯度质量浓度的标准品溶液进行线性回归,质量浓度与吸光值之间呈现出良好的线性关系(图4),得出多糖的线性回归方程:y=0.052 3x-0.025 5,决定系数r2=0.999 3。

图4 多糖标准曲线Fig.4 Standard curve of polysaccharide
2.2.2方法学验证 精密度试验测得滇黄精6次吸光度的RSD值分别为0.15%;重复性试验测得7个时间点的滇黄精样品吸光度的RSD值为1.05%;稳定性试验测得6份滇黄精样品吸光度的的RSD值为0.65%。50%、100%及150%浓度的样品量、加入量、测得量及回收率见表8,最后计算3个不同比例加入量的黄精多糖含量的平均回收率为98.81%,RSD值为1.03 %,表明该方法的准确度良好。
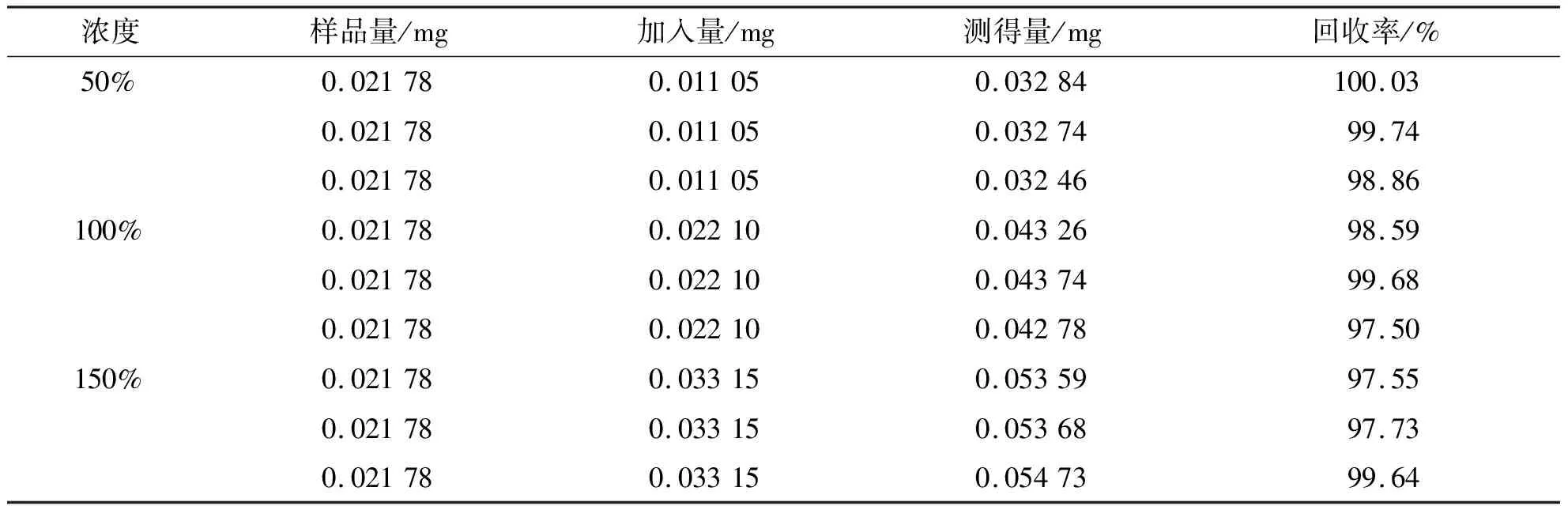
表8 滇黄精多糖的加样回收率Tab.8 Spiked recoveries of Polygonatum kingianum polysaccharide
2.2.3多糖含量测定 按照“1.2.5(1)”项下制备各批次供试品溶液,按照“1.2.5(6)”项下方法测定样品吸光度,9批滇黄精多糖含量为7.16%~27.27%(表9),均符合2020版《中国药典》(一部)规定的黄精多糖含量>7%的标准。该检测结果说明9个产地的黄精质量均较好,但不同产地间多糖含量存在一定差异,依次为贵州剑河>贵州安顺>贵州天柱>贵州铜仁>贵州赤水>贵州贵阳>贵州纳雍>广西百色>四川内江。

表9 不同产地滇黄精样品的多糖含量Tab.9 Polysaccharide content of Polygonatum kingianum samples from different origins
3 讨论
黄精作为药食两用大宗药材,用途广泛,始载于《名医别录》,其味甘、性平,具有补气养阴、健脾、润肺及益肾的功效,用于阴虚肺燥、阴血不足及脾胃虚弱之证[21]。据《中国植物志》记载,黄精属植物约60种,其中有20种为我国特有种,用途广泛,多做药用[22]。《中国药典》(2020版)收载黄精药材来源于百合科黄精属黄精、多花黄精及滇黄精3种原植物的干燥根状茎[1],其中滇黄精主要分布在云南、贵州、四川及广西等西南地区[22]。黄精属植物主要含多糖类、甾体皂苷类、黄酮类、三萜皂苷类、生物碱类及木脂素类等多种类型的化学成分[9]。其中甾体皂苷类成分(如薯蓣皂苷和原薯蓣皂苷)可通过降低胰岛素耐受等发挥降糖作用,且可通过抑制癌症细胞的增殖等发挥抗肿瘤作用[23]。三萜皂苷类成分(如羟基积雪草苷)在抗氧化、神经保护及抗肿瘤等方面具有广泛的药理作用[24];人参皂苷Rb1可通过提高胰岛素敏感性发挥降糖作用[25];黄酮类成分(如甲基麦冬二氢高异黄酮)不仅可通过抑制糖尿病大鼠糖基化终产物(advanced glycation end-product, AEG)的形成等发挥降糖作用,还可通过阻滞细胞周期等发挥抗肿瘤作用[26-27]。此外,芦丁、新甘草苷等黄酮类成分虽广泛存在于植物体中,但在抗氧化、抗糖尿病等方面具有重要作用,也可以作为该属植物发挥药效的基础物质[28-29]。
中药色谱指纹图谱具有“整体性”和“模糊性”两个基本属性,在物种区分、中药质量评价等方面具有明显优势,可从化学成分分布及含量分布比例等方面实现对同属不同物种、同一物种不同样品间的整体相似度进行比较[30]。本研究首先通过HPLC法构建了9批不同产地滇黄精指纹图谱,相似度评价良好,确认了12个共有峰;方法学考察精密度、重复性及稳定性良好(RSD<3%),提示此法准确可靠,可作为滇黄精药材的定性鉴定和质量评价的参考依据。9批不同产地来源的滇黄精指纹图谱相似度评价结果显示,除四川内江(S8)的相似度低于0.8,其余8批滇黄精药材的相似度均高于0.800,其中有7批药材来源于贵州,尤其是贵州纳雍(S3)、贵州安顺(S5)、贵州天柱(S10),相似系数均高于0.9,有效反映了贵州滇黄精均一性好、药材质量稳定。
多糖是黄精属植物的主要活性成分,是历版《中国药典》中黄精质量控制的指标。本研究参照2020版《中国药典》以蒽酮-硫酸法测定滇黄精多糖[1],9批滇黄精均符合药典标准,且黄精多糖含量均高于7%,其中含量高于20%的依次是贵州剑河、贵州安顺及贵州天柱,虽气候环境的不同会对黄精的质量产生一定影响,但实验结果显示贵州多数地区产的黄精质量略优于邻近省份。
通过指纹图谱共有峰的HCA结果显示,虽然欧氏距离>10时,四川内江、广西百色与贵州贵阳、贵州铜仁、贵州剑河存在交叉情况,但四川、广西两省的滇黄精多糖含量分别为7.16%和7.99%,而贵州各地区的黄精多糖含量均在12.67%及以上;主成分因子综合得分中贵州天柱产黄精最高,相较而言,贵州滇黄精整体质量较好,特别是贵州天柱、贵州赤水、贵州纳雍及贵州铜仁产地。
目前,黄精在农业、医疗保健、食品和化妆品等多个领域的开发都取得了显著的进展,市场对黄精的需求量巨大,以黄精作为主要原料的药品、食品及保健品种类繁多[31-32]。黄精药理活性显著,原料需求量正随着新产品的研发问世逐年增加,但黄精野生资源有限,已远远不能满足市场需求[33-34]。为扩大黄精产业未来发展,加强对黄精进行规范化种植,从源头保证黄精品质,深入探究不同产地、种属黄精及生态环境差异与质量间的关系,为全面健康的开发黄精产业资源提供支撑和参考依据。
综上所述,本研究通过建立滇黄精HPLC指纹图谱及黄精多糖含量测定,并采用聚类分析和主成分分析对9批滇黄精质量进行综合评价,不仅对黔产滇黄精的质量具有更加清晰的认识,研究结果也反映了贵州滇黄精均一性好,药材质量稳定,相较于邻近两省黔产滇黄精具有更优的质量,为贵州大宗药材的种植产业发展提供一定的科学依据。
