Active MoS2-based electrode for green ammonia synthesis
2024-04-22XinLiuLeiYangTaoWeiShanpingLiuBeibeiXiao
Xin Liu ,Lei Yang ,Tao Wei ,Shanping Liu ,Beibei Xiao ,,*
1 School of Energy and Power Engineering,Jiangsu University of Science and Technology,Zhenjiang 212003,China
2 Institute Charles Gerhardt Montpellier,UMR-5253,Université de Montpellier,CNRS,ENSCM,Place E.Bataillon,34095 Montpellier Cedex 05,France
Keywords: Nitrogen reduction reaction Density functional theory calculations Molybdenum disulfide Electrochemistry Catalyst Thermodynamics
ABSTRACT Nitrogen electro-reduction under mild conditions is one promising alternative approach of the energyconsuming Haber-Bosch process for the artificial ammonia synthesis.One critical aspect to unlocking this technology is to discover the catalysts with high selectivity and efficiency.In this work,the N2-to-NH3 conversion on the functional MoS2 is fully investigated by density functional theory calculations since the layered MoS2 provides the ideal platform for the elaborating copies of the nitrogenase found in nature,wherein the functionalization is achieved via basal-adsorption,basal-substitution or edge-substitution of transition metal elements.Our results reveal that the edge-functionalization is a feasible strategy for the activity promotion;however,the basal-adsorption and basal-substitution separately suffer from the electrochemical instability and the NRR inefficiency.Specifically,MoS2 functionalized via edge W-substitution exhibits an exceptional activity.The energetically favored reaction pathway is through the distal pathway and a limiting potential is less than 0.20 V.Overall,this work escalates the rational design of the high-effective catalysts for nitrogen fixation and provides the explanation why the predicated catalyst have a good performance,paving the guidance for the experiments.
1.Introduction
Artificial ammonia synthesis is of pressing concern for fertilizer production and hydrogen storage[1-4].In industry,the N2-to-NH3conversion is achieved through the Haber-Bosch reaction,which is highly energy consumption due to the inert N ≡N triple bond(941 kJ mol-1)[5-9].Such conventional method cannot satisfy the pursuit of energy sustainability.Thus,developing a green and clean technology to boost the nitrogen reduction reaction (NRR) is of paramount importance.In the direction,the ammonia synthesis via electrocatalysis under the mild conditions has triggered enormous investigations,wherein one critical hotpot is to discover the catalysts with high efficiency [10-15].
The marvelous nature provides an easy way to achieve the nitrogen fixation.For instance,Azotobacter endowed with nitrogenase enzymes enables the conversion from the atmospheric nitrogen to ammonia.As discovered,the nitrogenase is a two-comp onent system composed of the FeMo protein and the electrontransfer Fe protein.The FeMo protein contains the FeMo cofactor which provides the active site for the binding of reactant and reduction reactions[16].Inspired by the nature,counterfeiting welldesigned nitrogenase structure with inorganic structures made of similar elements and analogous configurations is an innovative method to design the catalysts for nitrogen fixations.Amongst varied materials,molybdenum dichalcogenide MoS2with layered configuration provides ideal platform for the elaborating copies of active site of natural nitrogenase[17-19].Therefore,there are great enthusiasms focused on MoS2in the application of NRR electro catalysis.
For the pristine MoS2,Mo-terminated edge has shown good performance in nitrogen fixation with a thermodynamic barrier of 0.68 eV [20];differently,the MoS2basal plane covered by the nonmetallic S atoms,is inactive for ammonia synthesis [20].To trigger its electrochemical activity,one vivid strategy is the heteroatom-adsorption on the basal plane.For example,Azofra et al.[16] deposited a single Fe atom on the basal plane and found an energy consumption of 1.02 eV.Zhang et al.[21] found that Fe2adsorption on MoS2has an overpotential of 0.21 V for NRR.Apart from Fe element,Zhai et al.[22]found that the Re decoration delivers the limiting potential of -0.43 V.Similarly,the transition metal substitution also offers the ability to activate the basal plane.Such as,the functional MoS2via Mo substitution has the limiting potential of -0.53 V [12].Despite all the breakthroughs,a full picture of the functional MoS2-based materials is still indispensable for further optimization the family of transition metal dichalcogenide.
To draw the whole map,we systematically investigated the N2-to-NH3conversion of the functional MoS2by means of the density functional theory calculations.Fig.1(a) illustrates the calculation models adopted in the work.For simplification,type I stands for the substitution at the Mo-terminated edge;type II and III are the adsorption and substitution on the basal planes,respectively.The elements of Tc,La and Hg are not taken into consideration due to the radioactive or toxic characteristic.In order to comprise the computation cost and the prediction accuracy,we do not perform the overall calculation progress and adopt the well-established descriptors.Fig.1(b) presents a four-step strategy to identify the active and stable candidates: 1) strong adsorption of N2molecule;2)low energy cost of the first and last protonation steps(the most likely limiting steps);3) relative high energy demanding of the competing hydrogen evolution reaction;4) thermodynamic stability under the electrochemical environment.Our results reveal that the edge-functionalization is a feasible strategy to boost activity.Theoretically,the Mo-terminated edge functionalized by single W1,dual W2or dual Re2is expected to catalyze the N2-to-NH3conversion under the overpotential of 0.16,0.19 or 0.25 V,being extremely attractive for the synthesis and application.This work provides the rational design of high-performance electrode for nitrogen fixation.

Fig.1.(a) The calculation models of the functional MoS2.TMn(I)is the substitution of transition metal at the Mo-terminated edge,TMn(II) is the adsorption of transition metal on the basal plane,and TMn(III)is the substitution of transition metal on the basal plane.The subscript n stands for the dopant number(single-atom or dual-atom).(b)The screening criterion of activity,selectivity and stability.
2.Computational Details
All calculations are performed within the density functional theory (DFT) framework as implemented in DMol3code [23,24].The generalized gradient approximation (GGA) with the Perdew,Burke,and Ernzerhof (PBE) functional is employed to describe the exchange-correlation interactions [25].The DFT Semi-core Pseudopotential (DSPP) is implemented for the relativistic effects of transition metals,which replaces core electrons by a single effective potential and introduces some degree of relativistic corrections into the core.The double numerical atomic orbital augmented by a polarization function (DNP) is chosen as the basis set [26].A smearing of 0.005 Ha(1 Ha=27.2114 eV)to the orbital occupation is applied to achieve accurate electronic convergence.In the geometry structural optimization,the convergence tolerances of energy,maximum force and displacement are 1.0×10-5Ha,0.02 Ha∙nm-1and 0.0005 nm,respectively.The spin-unrestricted method is used for all calculations.A conductor-like screening model(COSMO)was used to simulate the H2O solvent environment.COSMO is a continuum model in which the solute molecule forms a cavity within the dielectric continuum.The DMol3/COSMO method has been generalized to periodic boundary cases.The dielectric constant is set as 78.54 for H2O.The systems are fully relaxed for all the geometrical optimization.The type I catalysts were modeled on 4 × 3 × 1 supercell MoS2,and the type II and III models were built by using 3 × 3 × 1 supercell.A 1.5 nm-thick vacuum is added to avoid the artificial interactions between the catalyst and its images in the Z direction.The adsorption energy(Eads)is calculated by
where,Esystem,Ecatalystand Emrepresent the total energy of the adsorption system,the isolate catalyst and the adsorbates,respectively.
The binding energy(Eb)are calculated to examine the stability of all designed catalysts by
where,Evis the total energy of the catalyst without TM atoms,n is the number of TM atoms and ETMis the total energy of the single TM atom.
To evaluate the experimental feasibility of all designed catalysts,the formation energy (Ef) and dissolution potential (Udiss) are determined by
where,ETM-bulkis TM atoms energy in the corresponding bulk,(metal,bulk) and n are the standard dissolution potential of bulk metal and the number of electrons involved in the dissolution.The evaluation of dissolution potential is on basis of the previous works [27-30].
There are six proton-electron coupled steps involved in the electrochemical NH3synthesis from N2,i.e.,N2+6(H++e-)→2NH3.The Gibbs-free energy change (ΔG) of each elemental step was computed by standard hydrogen electrode model(SHE)raised by Nørskov et al.[31] wherein the chemical potential of(H++e-)pairs equaled to one-half of H2at standard condition [31].The ΔG was determined by
where,ΔE is the reaction energy analyzed directly from the DFT computations,ΔZPE and ΔS are the zero point-energy and the entropy difference at room temperature(T=298.15 K).The zero-point energies and entropies of the NRR intermediates are calculated from the vibrational frequencies.To reduce the calculation consumption,the substrates are fully constrained for the frequency calculation [32].ΔGUis the contribution of the applied electrode potential U and ΔGpHis the pH correction of the free energy expressed as ΔGpH=2.303×kBT×pH,where kBis the Boltzmann constant and pH is set to zero.ΔG<0 corresponds to an exothermic adsorption process vice versa.The methods have been successfully applied for analyzing N2fixation process [33,34],indicating its robustness and feasibility herein.Furthermore,the limiting potential was defined as the applied potential which can change each step into exergonic one.The limiting potential ULis determined by the following equation:
where,ΔGPLSis the free energy change of the step with maximum endothermic character.
3.Results and Discussion
The N2-to-NH3conversion is a complicated electrocatalysis coupled with successive proton-electron transfers.According to the Sabatier principle,the optimal activity needs the subtle balance between activation and poisoning [35].In principle,the stable surface would suffer from the first protonation step(*N2+H++e-→*NNH) meanwhile the reactive surface would limit by *NH2poisoning (*NH2+H++e-→*NH3) [36].Considering the low energy demanding for the*NNH formation and the*NH3formation are indispensable,the corresponding free energy changesare preferentially adopted as activity descriptors [22,36,37].Herein,a strict criterion of≤0.3 eV is artificially used which identify 17 potential configurations that may be highly active.Subsequently,we perform the full-path calculations on the promising systems to verify the potential-limiting steps(PLS)and obtain the limiting potentials UL.The free energy changes ΔG of elementary steps and the NRR thermodynamic barriers ΔGNRRare summarized in Table S1.The corresponding adsorption energies Eadsof NRR intermediates are listed in Table S2 for reference.As shown in Fig.S1 in Supplementary Material,there are the linear relations among the various Eadsdue to the similar adsorption configuration and the scaling between ULand adsorption ability,hinting the performances of candidates mentioned above are limited by the insufficient activation.
According to ULof 17 potential configurations summarized in Fig.2(a),we found 10 candidates with UL≤0.3 V,including W(I),W2(I),Re2(I),Fe2(I) and Ru2(I) as well as W(II),Nb(II),Zr(II),Fe2(II)and Mn2(II).The corresponding ULare 0.16,0.19,0.25,0.28 and 0.30 V for edge-substitution and 0.21,0.27,0.30,0.25 and 0.27 V for basal-adsorption,respectively.Our data on ULof Fe2(II) is in consistency with the study by Zhang et al.[21],which validates our calculation settings.Furthermore,we do not consider Fe2(I) in the following discussion owing to energetically unfavorable nitrogen adsorption.Generally,the dual-atom site provides enhanced activation capacity toward nitrogen molecule and is suggested to offer better activity in comparison with the single-atom site [38,39].However,it is not the case in the functional MoS2-based materials.Taken the basal adsorption as illustration,the single-atom sites of Zr(II),Nb(II) and W(II) have lower ULwith respective to the dualatom counterparts.
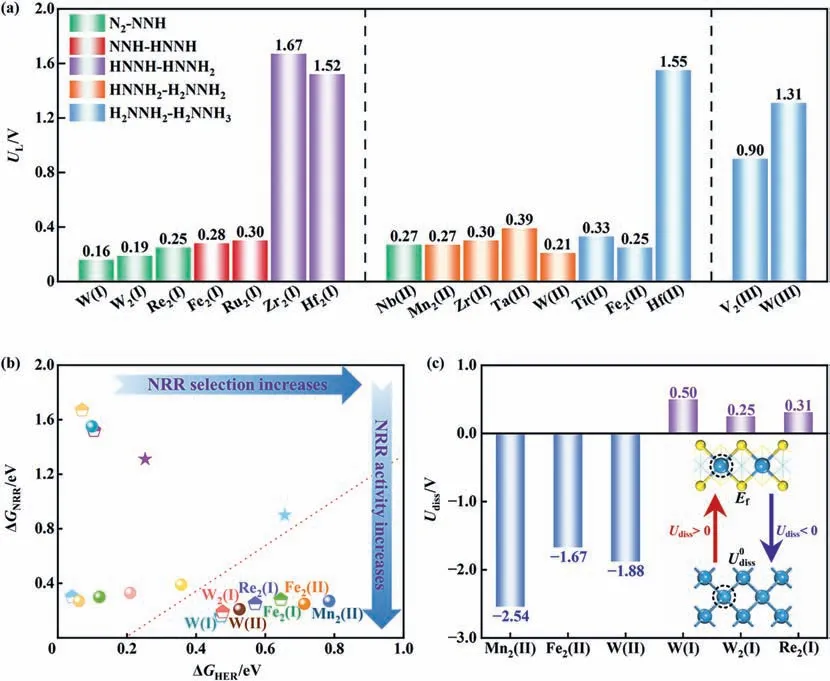
Fig.2.(a) The summary of the limiting potentials UL.(b) The thermodynamic barriers of nitrogen reduction reaction vs hydrogen evolution reaction (ΔGNRR vs ΔGHER).(c) The dissolution potential Udiss of the TM dopant.
In consideration of the catalysts might be also active in the reduction of protons into hydrogen under the same potential range,it leads to the competing hydrogen production [33,40,41].Therefore,in addition to the input energy to catalyze the ammonia synthesis,the selectivity of NRR vs.hydrogen evolution reaction(HER)should be considered.Herein,the selectivity is evaluated by the two parameters,that is,the comparison of adsorption energy Eads(*N2) vs.Eads(*H) and the comparison of thermodynamic barrier ΔGNRRvs ΔGHER.As listed in Table S2,Eads(*N2) are stronger than Eads(*H) for the candidates mentioned above,which ensures the preferential N2adsorption over H adsorption.Furthermore,we compare the thermodynamic barriers of NRR and HER (ΔGNRRvs.ΔGHER).Theoretically,it indicates a pretty good selectivity toward ammonia synthesis if ΔGNRR<ΔGHER.Fig.2(b)indicates that Ru2(I),Zr(II)and Nb(II)suffer from the limited selectivity and the efficient candidates are further narrowed to W(I),W2(I),and Re2(I)as well as W(II),Fe2(II),and Mn2(II).
Apart from the high efficiency,the structural stability is another important factor that needs to be considered.Since it is difficult to stimulate the structural evolution in the electrochemical environment,we simply adopt the dissolution potential Udissof the transition metal to give the information of the electrochemical stability under the applied potential [42,43],wherein the positive values indicate a resistance against electrochemical corrosion and the negative values mean de-alloying would occur by forming metal ions.The Udisspresented in Fig.2(c) reveals that edgefunctionalization is electrochemically stable but basal-adsorption would be degraded during the electrochemical cycles.The disparity stems from the interaction between the dopant and the surrounding coordination,as indicated by the binding energy Eblisted in Table S3.For instance,Ebare-9.56,-2.39 eV for W(I)and W(II),or -8.78 and -5.38 eV for W2(I) and W2(II),or -7.70 and -4.46 eV for Re2(I) and Re2(II).Therefore,the edge endowed with stronger Eboffers the significantly increased fixation ability toward dopant in comparison to the basal plane.Moreover,we performed the ab initio molecular dynamic stimulation at the temperature of 300 K to further ensure the thermodynamic stability of W(I),W2(I),and Re2(I).Fig.S2 clearly demonstrates the structural preservation of edge-functionalized configurations,which is also in linewith the strong binding.Moreover,due tothe possibility that the structural deformation caused by adsorbates,Fig.S3 presents the variation of TM-S bond lengths caused by the adsorption of NRR intermediates.Therein,the changes of bond lengths are within 0.015 nm and the TM-S bonds are always maintained without any collapse under the adsorption.Therefore,we identify robust W(I),W2(I),and Re2(I)for the N2-to-NH3conversion according to the fully consideration of activity,selectivity and stability.
Fig.3 demonstrates the free energy profiles of W(I),W2(I),and Re2(I)via the energetically preferred distal pathway meanwhile the alternative pathways and enzymatic pathways are presented in Fig.S4 and Fig.S5,respectively.The adsorption configurations in the insert demonstrate that the exposed edge provides flexible active sites,wherein the single-site is for the NNHxadsorption and the dual-site is accessible for NHxspecies.Under the potential U of 0 V,the *NNH formation is endothermic and the rest protonation steps are exothermic.In the regard,the identical PLS of *N2+H++e-→*NNH are observed for W(I),W2(I)and Re2(I)and the corresponding ΔGNRRare 0.16,0.19 and 0.25 eV,respectively.Furthermore,the free energy profiles of less stable W(II),Fe2(II)and Mn2(II) are provided in Fig.S6-S8 for reference due to the good efficiency in N2-to-NH3conversion.Wherein,the preferred reaction pathways are via enzymatic mechanism.The corresponding PLS are*HNNH2+H++e-→*H2NNH2for W(II)and Mn2(II)with ΔGNRRof 0.21 and 0.27 eV,and *H2NNH2+H++e-→*NH2for Fe2(II)with the ΔGNRRof 0.25 eV,respectively.The low ΔGNRRmatch well with the current records in the recent catalysts studies of the nitrogen electro-reduction [44-47].
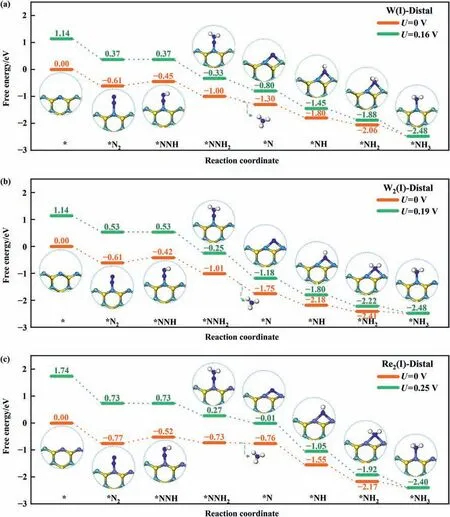
Fig.3.(a)The free energy profile of W(I)along the distal pathway.(b)The free energy profile of W2(I)along the distal pathway.(c)The free energy profile of Re2(I)along the distal pathway.Insets: the adsorption configurations.
It is worthy to note that there is a possibility of co-adsorption at the edge due to fully exposure of metal sites.In the regard,the influence of co-adsorption of N2molecule or H2O molecule on NRR performance is further investigated focused on the systems of W(I),W2(I) and Re2(I).The free energy profiles are given in Fig.S9,S10 and the corresponding ΔG of elementary steps are listed in Table S4.Under N2co-adsorption,the PLS is maintained at the first protonation step and ΔGNRRare slightly increased to 0.23,0.27 and 0.30 eV,respectively.It indicates the preservation of NRR reactivity.On the other hand,the spontaneous formation of *NNH2from*NNH occurs on W(I) and W2(I) due to the help of H2O molecule.The PLS of W2(I)changes to be the last protonation step with ΔGNRRof 0.09 eV and all ΔGiof W(I) are exothermic unexpectedly during the whole protonation steps.It means that the thermodynamic feasibility under the aid of H2O molecule.However,such phenomenon does not observe at Re2(I) wherein PLS is still located at the first protonation step and the ΔGNRRincreases to be 0.37 eV.Therefore,it should be realized that the solution environment poses impact on the NRR reactivity.However,we have to admit that the stimulation of solution environment is challenging which needs further exploration.
Based on the mentioned discussions,the candidates of W(I),W2(I),and Re2(I)are highly attractive for experimental preparation.By far we could tell that,the edge-functionalization is a feasible strategy to improve the MoS2activity since the basal-adsorption and basal-substitution show a low stability during the electrochemical cycles and low efficiency.Moreover,the exposed edge of the MoS2semiconductor has a good electron conductivity,benefitting for electron transfer in the field of electro-catalysis[48].As reported,Wang et al.[49]used one-pot heat treatment method to achieve different concentrations of W doping at the edge of MoS2.Similarly,Shi et al.[50] prepared different transition metal doped MoS2(TM=Fe,Co,Ni,Cu,and Zn)by one-pot solvothermal method.Therefore,it is not impossible task to synthesize the promising configurations we proposed in this work.
In order to uncover the origin of good performance,we perform the analysis on the electronic structures of W(I),W2(I),and Re2(I).Fig.4(a)shows the partial density of states between the p band of nitrogen molecules and the d band of W or Re.Therein,the strong pd orbital couplings are occurred around the Fermi energy level,indicating the formation of TM-N2bond.Furthermore,the right side of Fig.4(a)presents the charge density difference,wherein the electron accumulation on *N2reactant when adsorption on W(I),W2(I),and Re2(I),and the corresponding Mulliken charges Q are -0.075 e,-0.081 e and -0.052 e,respectively.As discussed previously,the N2-to-NH3conversion catalyzed by W(I),W2(I),and Re2(I) suffers from the first protonation step.The electrons accumulation on the *N2reactant could weaken the intermolecular N-N interaction and reduce the energetic upshift required for the *NNH formation [51,52].In order to further verify the electron transfer along the full-pathway,Fig.4(b) shows the Mulliken charges Q of NRR intermediates along the preferred distal pathway.The transformation from electron accumulation to electron depletion is observed when the reaction intermediate changes from *N2to *NNH2and from *N to *NH3.Clearly,there is electron communication between the edge and diverse intermediates during protonation steps,which is enabled by the TM d band cross the Fermi energy level as presented in Fig.S11.Generally,the W and Re active sites perform well in nitrogen fixation [22,27,45,53].For instance,ULare 0.29 or 0.39 V for W or Re anchored in g-CN[54],0.29 V for W embedded nitrogen-doped graphene[55],0.29 V for Re supported defective Mo2B2O2[56].It is well-known that once the molecule adsorbed,the metal site will transfer the electron to the molecules to activate the molecular orbitals and it will also accommodate the electron backward from the reactants,which is termed as the acceptance-back donation mechanism as described in Fig.4(c).This mechanism means it will be easy for the metal element with partial filling d band to activate the nitrogen molecules,which can explain why W and Re have shown good performances in our study since the electronic configurations are 5d46s2of W and 5d56s2of Re.

Fig.4.(a) The partial density of states between TM d band and *N2 p band for *N2 adsorption on W(I),W2(I),and Re2(I).Right-side: charge density difference.(b) The Mulliken charges Q of NRR intermediates along distal pathway for W(I),W2(I) and Re2(I).(c) The scheme of the acceptance-back donation mechanism.
As mentioned above,the optimal NRR performance needs the subtle balance between activation and poisoning [35],which directly relates to the affinity toward the intermediates of *N2and *NH2[36].Therefore,we further analyze the instinct factors that determine the good performance of the W/Re dopant for ammonia synthesis via charactering the key intermediates.According to the study by Fu et al.[57],the simple descriptor φ is calculated by,
where,Nd,χTM,and χsubare the d electron numbers of metal atoms,and the electronegativity of metal atoms and the electronegativity of substrates,respectively.The electronegativity of the substrate(χsub) is calculated by,
where,niand χiare the number and electronegativity of ith element,respectively.
Fig.5(a) indicates that the site with the larger φ offer stronger Eads(*N2).However,relative large deviation is observed for the linear fitting between φ and Eads(*NH2)in Fig.5(b),plausibly due to bridge adsorption manner.Furthermore,Fig.5(c),(d) demonstrates the reverse relationship between the descriptor φ and the Mulliken charge Q of the key intermediates mentioned above.The fittings hints the catalytic site endowed with the moderate φ values would offer good activity due to its ability to offer suitable activation and deliver feasible electron transfer.Therefore,the good performance of the N2-to-NH3conversion is expected for the W/Re dopant with φ values in the middle range.It is consistent with the acceptance-back donation mechanism.However,we have to admit that due to the limited data,it is not enough to capture the whole map and we will further work on edge-functionalization of TMS2and TMSe2.

Fig.5.Relationships between (a) adsorption energy of *N2,(b) adsorption energy of *NH2,(c) Mulliken charge on *N2 molecule Q(*N2),(d) Mulliken charge on *NH2 molecule Q(*NH2) and the descriptor φ.
4.Conclusions
In summary,we systematically investigated the functional MoS2as efficient N2fixation electrocatalysts.Our results demonstrate that the edge-functionalization is a feasible strategy to improve the activity of catalyst since the basal-adsorption and basalsubstitution suffer from the electrochemical instability and the NRR inefficiency.The robust edge-functionalization with W(I),W2(I),and Re2(I) configurations have shown good performances toward the N2-to-NH3conversion with high efficiency and structural stability.Overall,this work not only offers a comprehensive understanding of the stability,activity and selectivity of MoS2-based electrocatalysts,but also provides an effective strategy for designing the family of two-dimensional chalcogenides.
Declaration of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Acknowledgements
The authors greatly acknowledge the financial support from the National Natural Science Foundation of China(21503097,52130101,51701152,21806023,and 51702345)and China Scholarship Council(202008320215).
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2023.06.027.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Preparation of PrFexCo1-xO3/Mt catalyst and study on degradation of 2-hydroxybenzoic acid wastewater by catalytic wet peroxide oxidation
- Rational surface charge engineering of haloalkane dehalogenase for boosting the enzymatic performance in organic solvent solutions
- Long-term operation optimization of circulating cooling water systems under fouling conditions
- Effect of bubble morphology and behavior on power consumption in non-Newtonian fluids’ aeration process
- Preparation and properties of high-energy-density aluminum/boroncontaining gelled fuels
- Highly selective extraction of aromatics from aliphatics by using metal chloride-based ionic liquids