Green synthesis of ZSM-5 using silica fume and catalytic co-cracking of lignin and plastics for production of monocyclic aromatics
2024-04-22HongbingFuYufeiGuTianhuaGaoFuweiLiHengshuoGuHuchengGeYukeLiuZhixiaLiHongfeiLinJiangfeiCao
Hongbing Fu ,Yufei Gu ,Tianhua Gao ,Fuwei Li ,Hengshuo Gu ,Hucheng Ge ,Yuke Liu,Zhixia Li,*,Hongfei Lin,Jiangfei Cao
1 School of Chemistry and Chemical Engineering,Guangxi University,Nanning 530004,China
2 Guangxi Bossco Environmental Protection Technology Co.,Ltd.,Nanning 530007,China
3 School of Environmental and Chemical Engineering,Zhaoqing University,Zhaoqing 526000,China
Keywords: Silica fume ZSM-5 Catalytic co-cracking Plastics Lignin
ABSTRACT ZSM-5 with hierarchical pore structure was synthesized by a simple two-step hydrothermal crystallization from silica fume without using any organic ammonium templates.The synthesized ZSM-5 were oval shaped particles with a particle size about 2.0 μm and weak acid-dominated with proper Brønsted (B)and Lewis (L) acid sites.The ZSM-5 was used for catalytic co-cracking of n-octane and guaiacol,lowdensity polyethylene (LDPE) and alkali lignin (AL)to enhance the production of benzene,toluene,ethylbenzene and xylene (BTEX).The most significant synergistic effect occurred at n-octane/guaiacol at 1:1 and LDPE/AL at 1:3,under the condition,the achieved BTEX selectivity were 24% and 33% (mass) higher than the calculated values (weighted average).The highest BTEX selectivity reached 88.5%,which was 3.7% and 54.2% higher than those from individual cracking LDPE and AL.The synthesized ZSM-5 exhibited superior catalytic performance compared to the commercial ZSM-5,indicating potential application prospect.
1.Introduction
Monocyclic aromatic hydrocarbons (MAHs) such as benzene,toluene,ethylbenzene and xylene (BTEX) are important industrial chemicals,and are widely used in the production of chemical fiber,plastic and rubber [1,2].By far,the production of BTEX mainly depends on petroleum resourcesvianaphtha catalytic reforming and pyrolysis gasoline hydrogenation approaches [3,4].However,the increasing shortage of oil resource and demand for BTEX necessitates developing an alternative source to produce BTEX.As the sole renewable carbon resource,biomass has attracted much attention of researchers [5,6].Strenuous efforts have been made to produce BTEXviarapid pyrolysis and catalytic cracking of biomass [7,8].Nevertheless,due to the complicated chemical structure and high oxygen content (35%-50% (mass)) in biomass,the rapid deactivation of catalysts due to coking and low selectivity of target products could be the bottleneck problem countered in the large-scale application.Catalytic cracking of biomass are prone to produce toxic polycyclic aromatic hydrocarbons (PAHs) and phenolics [9,10].To resolve this issue,many studies carried out catalytic co-cracking of a co-feedstock of biomass and plastics (as a hydrogen-rich material),demonstrating a notable effect on enhancing BTEX production compared to the individual cracking of biomass [11,12].
It is well known that biomass consists of cellulose,hemicellulose and lignin.Every year about 50 million tons of lignin waste residue is produced from pulp engineering,which becomes a cheap under-utilized raw material [13].Pyrolysis and co-pyrolysis are suitable for lignin conversion due to its highly complex and recalcitrant features.Even though pyrolysis of lignin is difficult (due to easily forming coke)but lignin has the highest theoretical value to obtain BTEX due to its rich phenyl in its chemical structure.Therefore,a lot of research work for improving BTEX production were conducted on catalytic co-cracking of lignin and plastics.Zhanget al.[14] conducted catalytic co-cracking of black-liquor lignin and different plastics,found that co-cracking with polystyrene produced the maximum aromatics yield (55.3%),while co-cracking with polyethylene produced the maximum olefin yield (13%).Buet al.[15] reported that the addition of low-density polyethylene(LDPE) into the pyrolysis process of microwave-torrefied lignin enhanced aromatic hydrocarbon production (yield increased from 1.94% to 22.83%),promoted thermal degradation of lignin and improved the reaction rate.Development of novel catalysts and research on pyrolysis process for lignin are now in full swing.
ZSM-5 tends to be common catalyst used in the catalytic cracking of biomass due to its uniform micropores,tunable acidity and high hydrothermal stability[16].However,ZSM-5 is generally prepared by pure chemicals,e.g.tetraethyl orthosilicate and sodium silicate as the silica sources,and aluminum sulfate and sodium aluminate as the alumina sources,as well as organic structuring agent(OSDA).This increases the manufacturing cost and causes environmental pollution (due to the released toxic waste during the removal process of templates).Hence,it is necessary to find a ZSM-5 synthesis method with low-cost materials and without using an organic template.New synthesis pathway of ZSM-5 have been developed using low-cost fly ash,kaolin and rice husk ash[17–19].However,these methods could not avoid the use of the expensive OSDA [20,21].
Silica fume(SF)is a kind of primary industrial solid waste from the smelting process during the production of elemental silicon and ferrosilicon alloys [22].SF is a largely amorphous form of silicon dioxide,and typically exhibits a light weight,large specific surface area,fine grained(particle size<1 μm)and high activity.It is difficult to deposit and treat,and can’t be used efficiently.This results in the waste of resources and environmental pollution.At present,SF is mainly applied in concrete and refractory materials[23,24].Only a few of studies were carried out to use SF to produce mesoporous silicate materials[25]and microporous zeolite SSZ-13[26].The potential of SF as the silica source to synthesize zeolite catalysts needs to be explored in depth.
In the present work,ZSM-5 zeolite was prepared using SF as silica sourceviaa simple two-step crystallization method without using any OSDA.The catalytic performance was evaluated by cocatalytic cracking of LDPE and alkali lignin (AL),as well as their pyrolysis vapor model compounds (n-octane and guaiacol) to aim for enhancing the BTEX production from co-cracking of plastics and biomass.
2.Materials and Methods
2.1.Chemicals and pretreatment of SF
Sodium hydroxide (NaOH,≥96%),ammonium chloride (NH4Cl,≥99.5%),sodium sulfate (Na2SO4,≥99%) and sodium aluminate(NaAlO2,≥50%) were purchased from Guangdong Guanghua Sci-Tech Co.,Ltd,China.Hydrogen chloride (HCl,36%-38% (mass))was supplied by Chengdu Chron Chemicals Co.,Ltd,China.LDPE(MwandMnof LDPE are 93952 and 17898,respectively) was purchased from Shandong Shenhua Chemical Technology Co.,Ltd,China.AL,n-octane(99%)and guaiacol(99%)were purchased from Shanghai Macklin Biochemical Co.,Ltd,China.All chemicals were used as received.Commercial ZSM-5 (CZ,SiO2/Al2O3=27) was purchased from Nankai University Catalyst Co.,Ltd,China.SF was purchased from Henan Hengyuan New Material Co.,Ltd,China.
The chemical composition of SF analyzed by X-ray fluorescence spectrometry(XRF)is shown in Table 1.Virginal SF is mainly composed of SiO2with a small amount of Fe2O3,Al2O3and MgO,etc.In order to decrease the inhibitory action of the impurity in SF on the synthesis of ZSM-5,SF was washed with 10% (mass) HCl aqueous solution (at a SF/HCl mass ratio of 1:4) at 90 °C for 6 h under stirring.The acid-washed solid sample was filtered,washed,dried and calcinated at 550 °C for 6 h,and named as SF-A.As shown in Table 1,the Fe2O3content in SF-A obviously reduced,but the contents for other metal oxides slightly increased after acid washing.

Table 1 Chemical composition of SF and SF-A analyzed by XRF (% (mass))
2.2.Preparation of ZSM-5 from SF-A
ZSM-5 with a SiO2/Al2O3molar ratio of 30 was prepared by a two-step crystallization method.Firstly,4.56 g SF-A,0.5 g NaAlO2,and 0.1 g NaOH were dispersed in 10 g H2O and stirred for 1 h at room temperature.The mixture was transferred into a Teflonlined autoclave and maintained at 180 °C for 12 h (first step).Secondly,after cooling to room temperature,the obtained colloidal solid sample was mashed by using a pestle to form a smooth paste,and then mixed with a certain amount of NaOH and 20 g H2O,and further stirred for 1 h at room temperature.The resultant mixture was transferred into a Teflon-lined autoclave and crystallized at 180 °C for different times (second step).The obtained solid particles were filtered,washed and dried,followed by ion exchange twice with 1 mol∙L-1NH4Cl solution (at a solid/liquid ratio of 1/15 (g∙ml-1)) at 90 °C for 3 h.The obtained solid product was filtered,washed,dried and calcined at 550 °C for 6 h to obtain the SF-based ZSM-5 (SFZ).The samples were tabled,crushed and meshed to 20–40 mesh(0.425–0.850 mm)for subsequent catalytic cracking experiments.Here,the effects of the addition amounts of NaOH (x) and crystallization time (t) in the second step on the crystallinity of samples were investigated.The obtained samples were named as SFZ-xNaOH and SFZ-t,respectively.
2.3.Characterization of catalysts
The chemical composition of the samples was analyzed by XRF with an ARL Perform X4200W spectrometer.The crystalline characteristics of the samples were analyzed using a Rigaku D/max 2500v X-ray diffractometer (XRD).The microstructure of the samples was observed using a ZEISS Sigma 500 scanning electron microscope (SEM).The Fourier transform infrared spectroscopy (FTIR) measurement was carried out on a Nicolet 6700 spectrometer.N2adsorption–desorption experiments of all samples were carried out by a NOVA 2200e instrument.Prior to adsorption,all samples were degassed at 300 °C for 3 h.The Brunauer-Emmett-Teller (BET) surface area was calculated with the relative pressure ranging from 0.04 to 0.25.The total pore volume was derived from the adsorbed amount atP/P0=0.99.
The acidity of zeolites was analyzed by temperature programmed desorption of NH3(NH3-TPD),which was performed on an AMI-300Lite chemisorption apparatus.The detail about determination was revealed in our recent study [27].The types of acid sites on zeolites were analyzed byin situpyridine adsorption infrared spectroscopy (Py-IR) on a Tensor II FTIR spectrometer.The sample was pretreated at 300 °C under argon atmosphere for 2 h.After cooled to 30 °C,the pyridine was adsorbed for 10 min(pyridine was brought in using an argon flow at a flow rate of 10 ml∙min-1).The sample was heated up to the demand temperature and remained at this temperature for 5 min,and then the IR spectrum of the sample was recorded.
2.4.Catalytic cracking tests
Firstly,n-octane and guaiacol were selected as the model compounds in the pyrolysis vapor of LDPE and AL,respectively,and catalytically cracked in a fixed bed quartz tube reactor(inner diameter: 10 mm;length: 400 mm).In a typical run,0.3 g of catalysts was loaded into reactor,and heated to a desired temperature(450°C to 600°C)in N2stream(flow rate:30 ml∙min-1).The feedstock (0.3 g) was then injected into the reactor by a syringe pump(weight hourly space velocity (WHSV)=6 h-1) and reacted for 8 min.After flowing out from reactor,the cracked products were cooled at a cooling bath (-10 °C).The gaseous product was collected with the drainage method and analyzed according to the method described in our previous study [28].The liquid product was weighed and analyzed by GC–MS (gas chromatography -mass spectrometer,Agilent 5975C) and GC-FID (gas chromatography -flame ionization detector,GC-17A,Shimadzu).
In addition,LDPE and AL (the total mass: 0.3 g) were catalytically cracked to produce BTEX.In this case,solid samples were filled in a quartz basket,and slowly pushed into the reactor and reacted for 8 min at the given temperature.In order to improve the AL conversion and BTEX selectivity,catalytic cracking of cofeedstock of LDPE and AL was conducted at different LDPE/AL mass ratios (0:1,0.25:0.75,0.5:0.5,0.75:0.25 and 1:0).
The liquid yield (YL),solid residue yield (YSR),coke yield (Ycoke)and gas yield (YG) were calculated by the following equations.The relative content of specific component in liquid determined by the peak area normalization method was considered as the selectivity (Eq.(5)).The yield of each component was obtained by multiplying the selectivity with theYL(Eq.(6)).The hypothetical value of BTEX selectivity during co-cracking reaction was calculated by Eq.(7) [29].It is inferred that when the experimental value is higher than the calculated value,a synergistic effect between two feedstock occurs during co-cracking reaction.
whereMLis the mass of liquid product,M0is the mass of the raw material;MSRis the mass of solid residue;Mc0andMc1are the mass of the catalyst before and after reaction;xiis the area percentage of each component in the liquid determined by the peak area normalization method;SH-BTEXis the hypothetical value of BTEX selectivity,yis the mass fraction of LDPE in the co-feedstock;SLDPEandSALare the experimental BTEX selectivity when LDPE and AL are individually cracked.
The use stability of catalyst was determined by cracking the cofeedstock of LDPE and AL (LDPE/AL=0.5:0.5) for ten times (feed 0.3 g and react for 8 min every time) over the catalyst at 550 °C,followed by regenerating the catalystviacalcination in air atmosphere.Such ten times’ reactions-regeneration circles were repeated for three times.
3.Results and Discussion
3.1.Characterization of catalysts
3.1.1.Crystallization and morphology of samples
Both XRD patterns of SF-A and SF were similar and showed a broad amorphous feature(the figures are not shown here),demonstrating that SF-A and SF mainly contain amorphous silica.SEM images (Fig.S1 in Supplementary Material) showed that both SF and SF-A were spherical particles,and SF-A exhibited more smooth grain surface,indicating that those small impurity particles have been removed away by acid washing.
The XRD patterns of the samples undergoing the first step crystallization are shown in Fig.1(a).As can be seen,after 12 h’s crystallization,the sample (S1-12) exhibited slight characteristic peaks at 2θ=7.8°,8.8°,23.1°,23.7° and 24.2°,indicating the appearance of crystal seed of ZSM-5 (PDF-43-0321) [30].As seen in Fig.1(b),the diffraction peaks due to MFI structural characteristics became sharp and intense after the second step crystallization.SFZ-0.5NaOH achieved the highest crystallinity of ZSM-5 and no impurity peaks were observed,confirming that 0.5 g is the suitable addition amount for producing ZSM-5.When NaOH was added exceeding 0.6 g,the high alkalinity led to the formation of a more stable quartz phase [31].

Fig.1.XRD patterns of samples: the effect of crystallization time in the first step (a),different NaOH amounts (b) and crystallization time in the second step (c),(d) is the relative crystallinity of samples in the second step with extending crystallization time.
As shown in Fig.1(c) and (d),the relative crystallinity of samples (calculated based on the sum of peak areas between 2θ=7.8°–25° from XRD pattern of samples compared to that of commercial ZSM-5)increased significantly with extending crystallization time before 24 h,and then gradually increased after 24 h(the crystallinity was 65.8% at 24 h and 85.6% at 72 h).This could be due to the decreased available silica-alumina raw materials in crystallization liquid.
As shown in Fig.2(a)and(b),sample before crystallization reaction (S1-0) was spherical particles.After the first step crystallization reaction (S1-12),the interfaces between spherical SF particles became blurred,and these spherical particles trended to agglomerate into larger aggregates with some small particles adhering to the surface of larger aggregates.These results indicate that the original SF particles could be first corroded and softened,and then agglomerate into larger particles,accompanied by the formation of the crystal nucleus of ZSM-5 framework.In fact,the crystallization liquid became more and more sticky with extending crystallization time,consequently producing paste product after the first-step crystallization.

Fig.2.SEM images of samples from the first and second step crystallization,S1-x(the first step),x=0,12 h;SFZ-x(the second step),x=8,24,48,72 h,representing different crystallization times.
Fig.2(c)–(f) show the SEM images of samples from the second crystallization step for different crystallization times.As can be seen,a core–shell structured oval shaped particles were formed during crystallization process.The activated SF gel particles could compose the core,and the newly formed aggregates of the small crystals compose the shell.With extending crystallization time,the core disappeared gradually and the shell grew more complete,consequently leading to the formation of oval shaped particles with diameter of about 2.0 μm.
Fig.3(a) shows FTIR spectra of samples from the first and second step crystallizations.The absorption bands at 450 cm-1,790 cm-1,1100 cm-1result from Si—O—Si bending vibration,T—O stretching vibration (T represents Al or Si),and Si—O—Si asymmetric stretching vibration,respectively.The bands at 3620 cm-1and 3721 cm-1are attributed to the vibrations of framework bridging OH groups (Si—OH—Al) and the terminal Si—OH groups,respectively [32,33].The bands at 450 cm-1and 3620 cm-1are observed in S1-12 and SFZ-24,but absent in the initial mixture of materials(S1-0),indicating that the crystal nucleus precursor of aluminum silicate zeolites have been produced during the first step crystallization [34].The band at 550 cm-1and 1221 cm-1are identified as the double five-membered ring vibration of ZSM-5 framework and the asymmetric stretching vibration of four chains of the five-membered ring structure,respectively[32,35].The results confirmed the MFI crystal structure of the prepared zeolites.

Fig.3.FTIR spectra (a) and 27Al MAS NMR spectra (b) of samples after the first and second step crystallization.
The27Al MAS NMR spectroscopy was measured to analyze the environment of Al species in zeolites.As can be seen in Fig.3(b),S1-0 displayed two sharp peaks at 54 (framework tetrahedral Al species,AlFW) and around 7 (extra-framework octahedral Al species,AlEFW),while SFZ-24 showed only a sharp peak at 54,demonstrating that the most of AlEFW have been transferred into AlFW during crystallization process.
Combining the above analysis,a possible route for the formation of ZSM-5viathe simple two-step crystallization method is proposed in Fig.4.In the first step,the SF and sodium aluminate were firstly hydrolyzed,and then condensation reaction occurred between the resultant Si—OH and Al—OH groups to produce a viscous silica—alumina gel containing Si—O—T(T=Si or Al)structure.Crystal nucleus precursor of zeolite could be formed in this stage,as evidenced by formation of the five-membered ring of ZSM-5 from FTIR analysis.In the second step,the gel precursor was ground and mixed with a certain amount of NaOH aqueous solution,a colloidal suspension was produced.Afterwards,ZSM-5 framework was gradually formedvia in situself-assembly on the surface of colloidal particles with extending crystallization time.As observed in SEM images,the particles size at 8 h,24 h and 48 h were almost similar,and the newly formed small crystals aggregating on the surface of colloidal particles,therefore,we inferred that the crystallization reaction likely follows anin-situsolid hydrogel transformation mechanism [36].The presence of Na+helped to form the primary aluminosilicate species of fivemembered structures in the gel particles,and subsequently facilitate the condensation or rearrangement of the primary structure building units,consequently leading to the formation of spherical ZSM-5 particles which are actually aggregates of small cubic crystals.

Fig.4.Possible pathways for the synthesis of SFZ zeolites.
3.1.2.Acidity analysis
NH3-TPD profiles of samples are shown in Fig.S2.All samples showed two desorption peaks centered about 230 °C and 450 °C,which are generally attributed to the adsorbed ammonia on weak and strong acid sites,respectively [37].The acidity amount and acidity strength distribution were obtained by integrating the two peak areas.As can be seen from Table 2,all samples were weak acids-dominated materials,and the total acid amount increased,companied by the decrease of SiO2/Al2O3ratio of the samples with extending the crystallization time.These results indicate that more aluminum atoms entered the framework of zeolites with extending crystallization reaction,and thus produced more acidic sites[38].
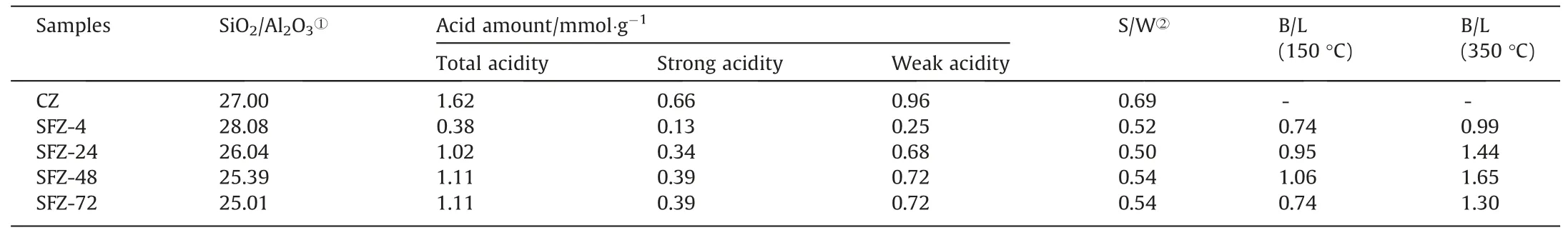
Table 2 Acid property of samples from different crystallization times.
Py-IR spectra from different samples are shown in Fig.5.The bands around 1537 cm-1and 1448 cm-1are attributed to the adsorption of pyridine on the Brønsted(B)and Lewis(L)acid sites,respectively[39].The band at 1484 cm-1is due to the vibration of the pyridine ring on both the B and L acid sites.As can be seen,both bands for B and L acid sites increased with extending crystallization time,indicating that the crystallization degree of samples was improved.However,the band intensity for L acid significantly decreased after desorption at 350 °C while the intensity of B acid sites remained unchanged,indicating that the B acid sites are mainly responsible for the strong acidic sites [40].The B/L value of samples (calculated by integrating the peak area of the adsorption bands) desorbed at 150 °C and 350 °C were 0.74–1.06 and 0.99–1.65 (Table 2),indicating that the as-prepared ZSM-5 is a weak acid-dominated catalyst with abundant B and L acid sites.

Fig.5.Py-IR spectra of samples desorbed at 150 °C (a) and 350 °C (b).
3.1.3.Textural properties of zeolites
The N2adsorption/desorption isotherm curves are shown in Fig.6(a).As can be seen,the CZ was typical I-type isotherms without any hysteresis loop even at a high relative pressure,indicating that the CZ is micropore-dominant material.The synthesized zeolites were IV-type isotherms with a H4 hysteresis loop at 0.45P/P0,indicating that the synthesized zeolites are a micro-and mesopores coexisting structure.In addition,the absorption isotherms indicate that the mesopores are connected to the micropores surface,causing capillary condensation effect on the intracrystalline channels and surface of zeolite[41].Fig.6(b)showed the pore size distribution of the synthesized zeolites,centered at about 2.5 nm with a small amount of mesopores about 11 nm.As shown in Table 3,the specific surface area (SBET),the mesopores volume(Vmes)and average pore diameters(Daver)of the samples increased with increasing crystallization time.These could be attributed to the growth of crystals and the aggregation of small crystals,leading to forming more intra-crystals microspores and inter-crystals voids (mesopores).
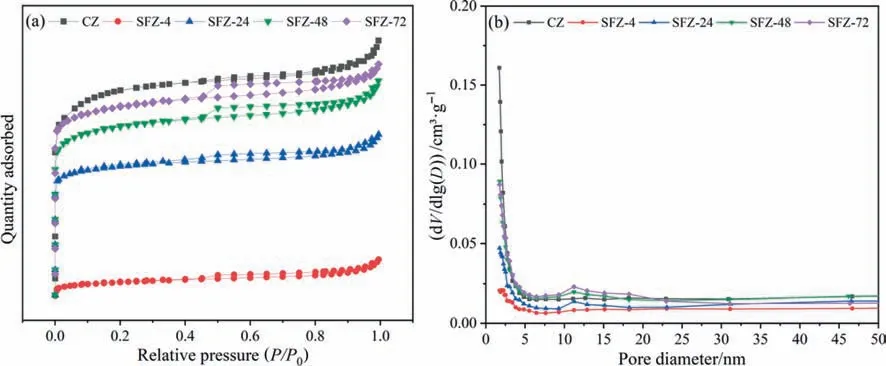
Fig.6.N2 adsorption–desorption isotherms (a) and pore size distributions (b) of the as-synthesized zeolites.

Table 3 Textural properties of the samples from N2 adsorption–desorption experiments
3.2.Catalytic performance
3.2.1.Catalytic cracking of n-octane and guaiacol
n-Octane and guaiacol were selected as the model compounds in pyrolysis vapor of LDPE and lignin,respectively,and cracked at different temperatures on SFZ-48 (with the highest acid amount).As shown in Fig.7(a),TheYLfrom crackingn-octane retained around 10% at the tested temperature range from 450 to 600 °C,while the BTEX selectivity significantly increased with the rise of reaction temperature and reached the maximal value at 600 °C (85.1%).The BTEX products at 600 °C contained toluene(45.3%),m,p-xylene (24.4%),benzene (6.0%),o-xylene (7.0%) and ethylbenzene (2.5%).A high temperature about 550–600 °C is favorable for producing BTEX from crackingn-octane.

Fig.7.The product yields and selectivity from catalytic cracking of n-octane (a,c) and guaiacol (b,d) at different temperatures.
As shown in Fig.7(b),the liquid products from cracking guaiacol mainly consisted of indene,phenols and naphthalenes with a small amount of benzene and toluene.The phenols decreased with increasing reaction temperature,accompanied by a significant increase in the selectivity to polycyclic aromatic hydrocarbons(PAHs)e.g.naphthalenes and indene.Even though the selectivity to benzene and toluene was improved at a higher temperature,however,the high temperature led to a lowerYLand a high PAH selectivity,therefore,550 °C could be the ideal temperature for producing BTEX from cracking guaiacol.
The gas products from cracking both model compounds mainly contained light alkanes (C1-4) and light olefins ().However,crackingn-octane obtained more propane while cracking guaiacol produced more methane.The light olefins selectivity from both model compounds increased with the rise of temperature,and reached the maximal value at 600 °C,36.4% forn-octane and 40.9% for guaiacol (Fig.7(c) and (d)),respectively.These light olefins are essential intermediate products to form aromatic hydrocarbonsviaDiels-Alder reaction [42].
Based on the above analysis on gas and liquid products,it was considered that the product distribution is strongly dependent on the chemical structure of raw materials.Crackingn-octane on the acid sites of catalysts can produce C1-C7fragments including light alkanes and alkenes [43].Especially due to the low bond energy the middle C—C bond in then-octane long chain can be easily broken at a high temperature,consequently leading to the formation of more propane and propylene.BTEX can be producedviathe direct cyclization of C6-C7,followed by dehydrogenation reaction,and alsoviaoligomerization and Diels-Alder reaction from light olefins [44].
The methyl and methoxy groups of guaiacol are easily removed at a high temperature,consequently producing methane in gas product and phenols [45].Phenols further react with methyl or alkyl radicals to form alkylphenols (e.g.2,4-xylenol,2-propylphenol).Meanwhile,phenyl radicals could be produced by deoxygenation reaction (or dehydration) of phenols,and afterwards probably transfer into benzene and toluene after combiningactive hydrogen or methyl radicals.Naphthalenes are formed by the coupled reaction of phenyl radicals [46,47].The formation of a lot of indenes could be attributed to the oligomerization of benzene with light olefins.
In addition,catalytic cracking of co-feedstock ofn-octane and guaiacol were carried out at 550°C.As can be seen in Fig.8(a),with increasing then-octane/guaiacol mass ratios (O/G ratio),the BTEX selectivity,especially toluene andm,p-xylene,significantly increased,while theYLand the selectivity to PAHs (e.g.indene and naphthalenes) substantially decreased.As shown in Fig.8(b),the propane selectivity obviously increased but those of methane,propylene and ethylene significantly decreased with increasing the O/G ratio.Meanwhile,theYGcontinued to increase with the rise of O/G ratio.These results indicate that the presence ofn-octane significantly accelerated the formation of gas products (especially propane) and promoted the BTEX selectivity in liquid products.This result was consistent with those from individual cracking ofn-octane and guaiacol.
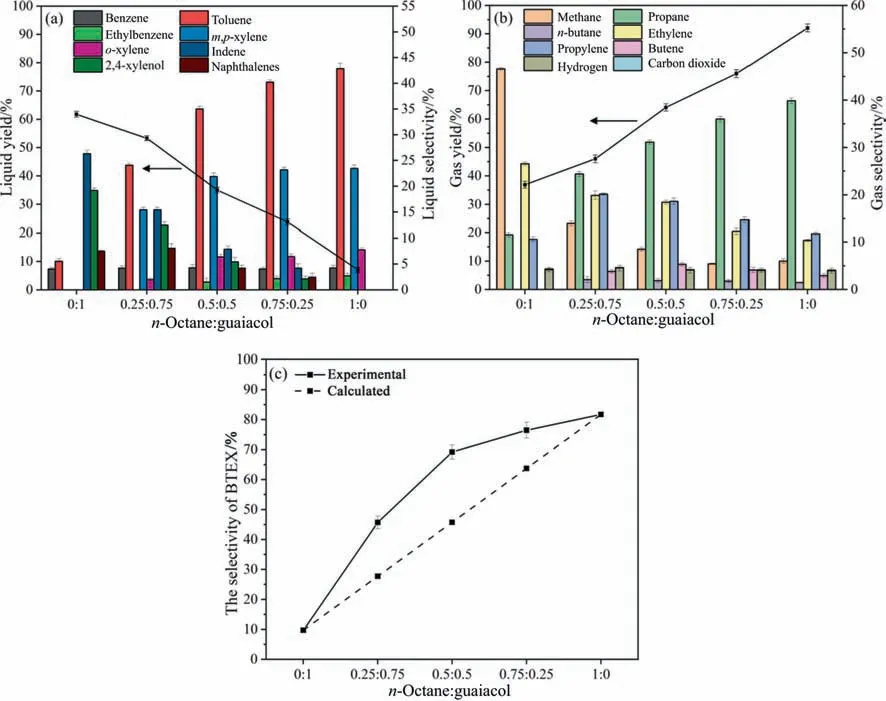
Fig.8.The yields and selectivity of liquid (a) and gas (b) and a comparison between the experimental and calculated BTEX selectivity (c) from catalytic co-cracking of n-octane and guaiacol.
Fig.8(c)shows a comparison between the calculated values(according to the Eq.(7)) and the experimental values of BTEX selectivity at different O/G ratios.The experimental values were always higher than the calculated values,and the largest difference between two values(24%)occurred at the O/G ratio of 0.5:0.5,indicating that two materials have significant synergistic effect on enhancing BTEX production during co-cracking process.The synergistic effect could be attributed to several paths (Fig.9): (i) crackingn-octane can produce C6-C7fragments,which can be transferred into BTEXviadirect cyclization and dehydrogenation reaction.(ii) crackingn-octane can also produce a large number of light olefins,which form a hydrocarbon pool,and thus facilitate the Diels-Alder reaction,consequently leading to the formation of BTEX[48];(iii)guaiacol tends to transfer into phenol and catechol after undergoing demethoxylation and demethylation reactions,these phenols can further convert into phenyl radicals after undergoing deoxygenation reaction.These phenyl radicals could combine with the methyl or ethyl groups (cracking intermediates fromn-octane) to produce toluene,xylene and ethylbenzene[46],and/or transfer into benzene by combining active H radicals.Besides,PAHse.g.naphthalenes (as undesired products) can be formedviacoupled reaction of phenyl radicals.

Fig.9.Possible reaction mechanisms of cracking the co-feedstock of guaiacol and n-octane.
3.2.2.Catalytic cracking of LDPE and AL
Lignin is the most thermal stable and has the highest theoretical yield to obtain BTEX among the three main components of biomass.Here,cracking of alkali lignin (AL),LDPE and co-feedstock of AL and LDPE were carried out to enhance the production of BTEX.As shown in Fig.10(a),theYLfrom cracking LDPE at the tested temperature range varied from 11.6% to 22.0%,and the highest BTEX selectivity was 86.7% achieved at 550 °C,which included toluene (43.5%),m,p-xylene (26.2%),benzene (7.1%),ethylbenzene(3.1%)ando-xylene(5.2%).TheYLfrom LDPE was obviously higher than that fromn-octane(model compound),which could be due to the pyrolysis vapor (with larger molecular) from LDPE merely undergoing preliminary cracking on zeolites in a limited time,consequently avoiding further cracking reaction to produce gas.However,the BTEX selectivity was similar to that of then-octane,indicating that LDPE andn-octane follow similar cracking reaction mechanism.
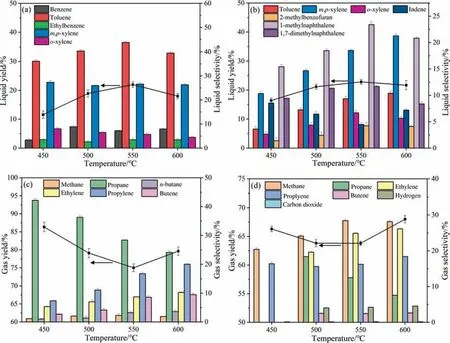
Fig.10.The product yields and selectivity from catalytic cracking of LDPE (a,c) and AL (b,d) at different temperatures.
The liquid product from individually cracking AL mainly contained naphthalenes,m,p-xylene,toluene,o-xylene,indene and 2-methylbenzofuran(Fig.10(b)).TheYLand BTEX selectivity were obviously improved by increasing reaction temperature,however,the high temperature(550–600°C)resulted in the increase of PAHs such as naphthalenes and indenes.Compared to cracking guaiacol(the main product were indenes 30% and phenols 20%),cracking AL obtained a lowerYL(22% at 550°C)but produced more naphthalenes (e.g.35% at 550 °C) and xylenes (e.g.27% at 600 °C),which could be attributed to the difference on the chemical structure of feedstocks.The side chains of benzene ring of guaiacol are methoxy and hydroxyl groups,it is easy to form stable phenols (e.g.2,4-dimethylphenol,2-propyl phenol)viademethoxylation and demethylation reactions.AL is a composite of dimer,trimer and even polymer of phenyl propane,and thus during the cracking process AL is firstly pyrolyzed into vapor,the vapor is then catalytically converted on the active sites of zeolites.A large amount of phenyl and methyl radicals in pyrolysis vapor are active and easily combine with each other to form methylnaphathalene,dimethylnaphathalene,toluene and xylenes.
As can be seen in Fig.10,for both feedstock,the gas yields are relatively lower while BTEX selectivity are relatively higher at 550–600 °C,indicating the 550–600 °C is the ideal temperature range to obtain liquid producte.g.BTEX.Propane selectivity significantly decreased while that of ethylene increased with increasing temperature,indicating that propane trends to crack into ethylene at a high temperature.More propane and propylene were present in the gas product from cracking AL compared to crack guaiacol,indicating that gas products mainly derive from the breakage of side chains of phenyl propane structural units.
Catalytic co-cracking of LDPE and AL was carried out at 550 °C,and the product distribution from cracking the co-feedstock is shown in Fig.11.As can be seen,theYLremain at about 20% at any raw material ratios.The BTEX selectivity significantly increased while the selectivity to naphthalenes obviously decreased with increasing the LDPE/AL ratio,indicating that adding a certain amount of LDPE in feedstock significantly restrains the formation of the undesirable PAHs,but enhances producing valuable MAHs.The highest BTEX selectivity (88.5%) was achieved at a LDPE/AL ratio of 0.5:0.5,which was 3.7% higher than cracking of LDPE and 54.2% higher than cracking of AL.The experimental BTEX selectivity from cracking of co-feedstock were always higher than those calculated values (Fig.11(c)),indicating that LDPE and AL have a strong synergistic effect on enhancing the BTEX production during co-cracking process [11,49].

Fig.11.The yield and selectivity of liquid(a)and gas(b)products and a comparison between experimental and calculated BTEX selectivity from co-cracking of LDPE and AL(c).
The products composition from cracking LDPE,AL and cofeedstock at 550 °C is shown in Table S1.Cracking AL mainly formed naphathalenes (including 1-methylnaphthalene and 2,7-dimethylnaphthalene),m,p-xylene and toluene.The cracking products from LDPE and co-feedstock were similar,and mainly included toluene,m,p-xylene and benzene.This indicates that the formation of naphthalenes is restrained by mixing LDPE with AL.By combining our studies with data published in literatures,the reaction mechanism for catalytic co-cracking of AL and LDPE is proposed in Fig.12.At a high temperature,the ether bonds in the AL break to form a large number of phenoxy and benzene radicals while release methyl and methoxy radicals (Route 1);these unstable radicals combine with each other to produce the desired componentse.g.toluene and xylene(Route 2)as well as the undesired componentse.g.naphthalenes (Route 3).Cracking LDPE produces a large amount of long and short-chain alkanes and olefins(Route 4).The long chain alkanes and olefins above C6can be directly cyclized and dehydrogenated to produce aromatics(Route 5)when they pass through the catalyst bed.The short olefins form“hydrocarbon pools”,which undergo oligomerization reaction and Diels-Alder reaction to grow the chains,followed by cyclization and dehydrogenation to produce aromatics (Route 6) [50,51].The large amount of alkyl radicals(from cracking of LDPE)can combine with phenyl radicals (from cracking of AL) to produce more toluene and xylene (Route 7),consequently preventing coupling reaction between phenyl radicals to form PAHse.g.naphthalenes and coke.
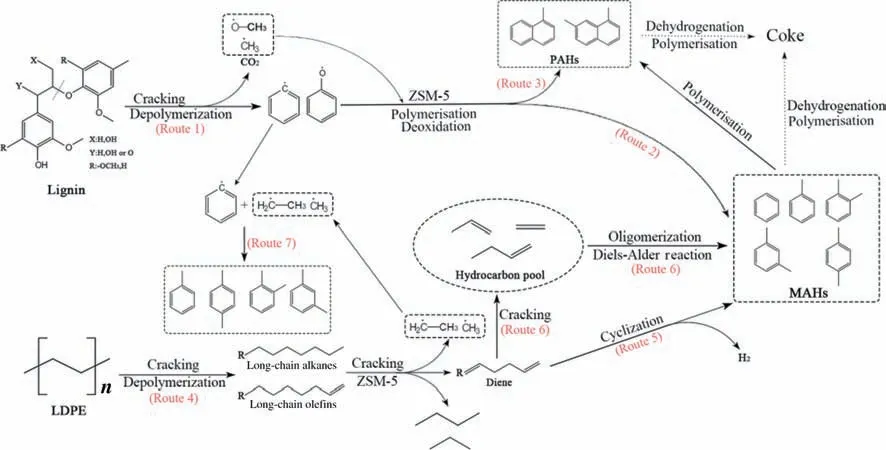
Fig.12.Possible reaction mechanism of co-catalytic cracking of LDPE with AL.
The catalytic performance of SFZ-48 was compared with that of CZ at 550°C.As can be seen in Fig.S3,the SFZ-48 achieved a higherYLfrom cracking co-feedstock,and a slight higher BTEX selectivity upon cracking three feedstocks compared to CZ.This could be attributed to the proper acidity and novel porous structure of SFZ-48.The weaker acidity and proper B and L acid sites distribution help to restrain coking of cracking vapor on the surface of catalyst,while the hierarchical micro-/meso-pores coexisting structure allows cracking vapor rapidly entering the micropores of catalyst,consequently promoting the formation of BTEX.We conducted control experimentviapyrolysis of the co-feedstock(LDPE/AL=0.5:0.5) with quartz (as heat-transfer medium) at 550 °C,and compared the cracking effect of different catalysts at different catalyst dosages (0.3 g,0.45 g and 0.6 g).As shown in the Fig.S4,the order of liquid yields for three catalyst dosages are SFZ-48 >CZ >quartz (for instance,13% for quartz,17.5% for CZ and 21% for SFZ-48 at the catalyst dosage of 0.3 g);except for quartz (BTEX selectivity remained about 73%),BTEX selectivity increased with increasing catalyst dosage,reaching 90.6% for CZ and 92.1% for SFZ-48 at a catalyst dosage of 0.6 g,which are 5.4% and 4.6% higher than those from 0.3 g.The results show that the increase of catalyst dosage improved the BTEX selectivity in product,and SFZ-48 showed superior catalyst performance than the commercial ZSM-5.In another study,alkaline immersing-treated ZSM-5(SiO2/Al2O3=35)were used for co-cracking of milled wood lignin and high density polyethylene with a fixed bed [52].The obtained BTEX selectivity was 71.8%,which was 15.7% lower than that achieved in present study(87.5%).These results prove that the prepared catalyst is comparable with the commercial ZSM-5,suggesting the potential application prospects.
3.2.3.The stability of catalyst
As shown in Fig.13(a),YLincreased with the rise of reaction cycles,but BTEX selectivity significantly decreased with increasing reaction times,especially in the third cycle.YGsignificantly decreased with increasing reaction times and cycles,accompanied with the increase of light olefins () selectivity regardless of CZ or SFZ-48.The averageYcokeof SFZ-48 and CZ after first,second and third cycles reached 9.9%,12.1%,15.3%,and 10.9%,13.8%,16.5%,respectively.The formation of more coke on CZ is mainly attributed to its strong acid sites[53].Such variation trends of yields and distributions of liquid and gas products could be attributed to the structural damage and increasing coke deposition (especially in the micropores) on catalysts,which lead to the decrease of catalytic activity on cracking and aromatization reactions.Besides,it is noteworthy that in each cycle,BTEX selectivity from SFZ-48 were always higher than those from CZ,suggesting the better catalytic performance of SFZ-48 for enhancing the production of BTEX.

Fig.13.The yields and distributions of liquid and gas products with SFZ-48 and CZ in three consecutive reactions-regeneration cycles.
After the first and second regeneration,BTEX selectivity recovered about 80% and 60% of the initial value,but continuously decreased as the reaction proceeded.The spent catalyst (after ten time’s consecutive reactions) was analyzed by NH3-TPD and N2adsorption–desorption experiments (Fig.S5,Table S3).The total acid amount of the used SFZ-48 dropped to 0.55 mmol∙g-1and was recovered to 0.89 mmol∙g-1after regeneration,which were lower than that of fresh SFZ-48 (1.11 mmol∙g-1).The specific surface area,pore volume and pore size of the used SFZ-48 decreased significantly after cracking reaction,but increased after regeneration.These results indicate that the use stability of catalyst needs to be further improvedviaoptimizing synthesizing process to promote the crystallization degree of zeolites.
4.Conclusions
ZSM-5 with hierarchical pore structure was synthesized by a simple two-step hydrothermal crystallization from silica fume without using any organic ammonium template.The resultant ZSM-5 were oval shaped particles with a particle size about 2.0 μm consisting of the aggregate of small crystals,and had proper acidity with abundant B and L acid sites.The resultant ZSM-5 was used for catalytic co-cracking ofn-octane and guaiacol,LDPE and AL to enhance the BTEX production.The most significant synergistic effect occurred atn-octane/guaiacol at 1:1 and LDPE/AL at 1:3,as analyzed that the experimental BTEX selectivity were 24% and 33% higher than the calculated values.The highest BTEX selectivity reached 88.5%,which was 3.7% and 54.2% higher than those from cracking LDPE and AL alone,respectively.The alkyl radicals(derived from cracking of LDPE) combine with phenyl radicals(from cracking of AL) to produce more toluene and xylene,which significantly contributes to the synergistic effect on enhancing the BTEX formation.Our study developed a green process to synthesize ZSM-5 from industrial solid waste and produce valuable fine chemicals from co-cracking of biomass and waste plastics.
Data Availability
Data will be made available on request.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078076),Guangxi Natural Science Foundation(2020GXNSFAA159174)and the Opening Project of National Enterprise Technology Center of Guangxi Bossco Environmental Protection Technology Co.,Ltd (GXU-BFY-2020-005).
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cjche.2023.07.006.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Flower-like tin oxide membranes with robust three-dimensional channels for efficient removal of iron ions from hydrogen peroxide
- Experimental study on the activation of coal gasification fly ash from industrial CFB gasifiers
- Enhanced stability of nitrogen-doped carbon-supported palladium catalyst for oxidative carbonylation of phenol
- Solubility of iron(III) and nickel(II) acetylacetonates in supercritical carbon dioxide
- Filtration performance and modeling of granular bed for dust removal from coal pyrolytic vapors
- Copper slag assisted coke reduction of phosphogypsum for sulphur dioxide preparation