HTPB/Al/AP/RDX推进剂初始燃烧的分子模拟
2024-04-19初庆钊付小龙郑学明刘金龙陈东平
初庆钊,付小龙,郑学明,刘金龙,陈东平
(1.北京理工大学 爆炸科学与技术国家重点实验室,北京 100081;2.西安近代化学研究所,陕西 西安 710065;3.黑龙江北方工具有限公司,黑龙江 牡丹江 157013)
引 言
HTPB推进剂是武器的重要动力来源,其不仅具有较宽的燃速调节范围且力学性能好,制造工艺简单,在国内外多种火箭发动机型号中得到应用[1,2]。HTPB推进剂成分包含不同级配的AP、RDX、Al颗粒物以及HTPB黏合剂等物质,是一种非均质复合含能材料。
为了研究HTPB推进剂的燃烧理论模型,Beckstead等[3,4]提出了BDP多火焰模型,将HTPB推进剂燃烧分为3个阶段,使用简化反应机理描述AP/HTPB的反应过程。颜密[5]采用BDP 微尺度燃烧模型对AP/HTPB复合推进剂进行模拟,并进行稳态计算和燃速验证,研究了工作压强、AP 浓度和 AP 粒径这3个参数对组分扩散特性、气相温度分布以及火焰位置的影响,为推进剂压力耦合响应函数的数值计算提供理论基础。张晏榕等[6]提出了一套气固耦合的三维空间求解数值框架,使用两套不同网格描述气相和固相演化,实现了三维非均质推进剂的燃烧模拟。目前的燃烧模拟研究大多关注AP/HTPB两种组分,对于HTPB/Al/AP/RDX四组元推进剂,其界面存在复杂的相互作用机制,相关基础研究缺失,导致难以开展燃烧模拟研究。
推进剂配方直接影响推进剂的物理化学性质,不同组分之间的界面上存在着复杂的相互作用,导致其燃烧、爆炸、安全性能等发生本质改变。国内外学者针对四组元HTPB推进剂的燃烧特性开展了基础实验研究。黄蒙等[7]对HTPB/Al/AP/RDX推进剂组分之间的相互作用开展研究,通过热分解动力学分析,发现在热分解阶段,组分HTPB、AP和RDX之间存在明显的相互作用。其中, AP会强烈加速HTPB和RDX的分解;反之,HTPB和RDX也会促进AP分解。吴迎春等[8]采用全息技术对HTPB/Al/AP推进剂燃烧过程中的颗粒行为进行可视化分析,发现铝颗粒燃烧过程中经历了剥落、微爆、燃烧等过程。尽管前人针对HTPB推进剂已开展了系列实验研究,对于推进剂组分间影响规律有一定的认识,然而推进剂组分间的相互作用往往发生于微纳米尺度,受限于实验手段和设备,尚未有合理的实验方法可直接测试其微观反应机制,这导致HTPB推进剂微观反应机制缺失,限制了其性能的优化提升。
分子动力学模拟可以深入探究微观物理化学机制,是研究复杂燃烧过程的重要工具,在复杂体系反应模拟中得到了广泛的应用[9-13]。其往往依赖于力场或势函数模型,目前含能材料领域中最著名的力场是van Duin 和Goddard提出的ReaxFF可反应力场[14],该方法通过考虑原子间键级来描述化学反应过程,已被应用于研究铝粉[11]、高能炸药[15]的燃烧与爆炸机理。在HTPB推进剂中,AP是重要的氧化剂组分。但是,目前还未有适用于AP反应体系的ReaxFF力场参数,这限制了AP热解与燃烧反应机理的研究。近期,本课题组针对AP单质开发了机器学习势函数[16],研究了AP热分解过程中的详细反应机理,观察到了AP受热过程中相态演化、吸/放热过程以及最终的气相产物。虽然,在过去20年间分子动力学力场在含能材料领域中取得了重大的成功,但是目前已公开的势函数模型通常仅能描述推进剂中1~2种组分,这无法满足推进剂全配方分子模拟的现实需求。推进剂全配方分子模拟一直是世界性的难题,不仅需要描述推进剂单质组分,还需要考虑组分间复杂的相互作用,目前国际上未有成功先例。如若能建立推进剂全配方的势函数模型,有望开展真实推进剂配方燃烧、力学等物理化学过程的全原子模拟,实现推进剂的理性设计。
基于以上分析,本研究针对HTPB/Al/AP/RDX等推进剂关键组分开发了一个通用势函数。该势函数基于第一性原理计算的数据集,使用深度神经网络模型进行开发。针对推进剂组分的能量、受力开展了测试验证。随后,建立了HTPB四组元推进剂燃面模型,并进行了大规模的分子动力学模拟计算。在模拟计算中,成功观察到了推进剂燃烧的相变、热解、传热、传质等微观过程。在世界范围内首次实现了HTPB四组元推进剂的燃烧微观模拟,揭示了其中的界面作用机制,为固体推进剂的理论建模提供了新的思路。
1 模拟方法
1.1 势函数开发
分子动力学模拟的核心是反应势函数模型,本研究采用深度势能框架(DeepMD)中的神经网络模型来拟合势函数参数[17]。图1为机器学习势函数开发流程。
图1 机器学习势函数开发流程Fig.1 Development process of machine learning potential function
在图1中,训练数据集由第一性原理MD模拟的轨迹组成。然后,建立了一个神经网络势函数(NNP)模型,该模型将原子配位(R)解释为原子间力(F)和能量(E),从而实现了在大空间和时间尺度上具有第一性原理精度的MD模拟。模型中每个结构的能量由结构内每个原子能量之和表征:
E=∑Ei
(1)
式中:E为结构能量;Ei为原子i的能量,采用公式(2)计算:
Ei=Ewɑi(Ri)
(2)
式中:αi表示原子i的化学元素;wαi表示原子i的神经网络参数;Ri为原子的环境矩阵。
(3)
式中:rij表示原子i、j间的笛卡尔坐标距离。
随后建立深度神经网络对结构的能量与受力进行预测。深度神经网络包含一个三层(25、50、100节点/层)的滤波(嵌入)网络和一个三层(240节点/层)的拟合网络。与经典神经网络相似,DeepMD方案通过使用反向传播算法计算损失函数的梯度来训练模型。NNP训练共经过4.0×106次迭代,学习率从1.0×10-3到5.0×10-8呈指数衰减。训练过程中的损失函数L为:
训练集包括HTPB/Al/AP/RDX等组分的单质以及任意两两组分间的界面结构。在300、1000、2000、3000和4000K的温度下,进行了一组1ps的第一性原理MD模拟,以获得NVT系综下的运动轨迹。第一性原理MD模拟使用CP2K[18]直接计算,计算方法采用Goedecker-Teter-Hutter (GTH)赝势和Perdew-Burke-Ernzerhof (PBE)广义梯度近似方法处理核心电子,基组考虑了双ζ高斯基函数加极化(DZVP-MOLOPT)。其他详细的设置请参考本课题组关于NNP开发的相应工作[19-21]。
1.2 四组元推进剂模型
图2为构建的HTPB/Al/AP/RDX四组元推进剂装填模型。

图2 HTPB/Al/AP/RDX四组元推进剂装填模型(图中的 C/H/N/O/Al/Cl 分别使用灰/白/蓝/红/银/绿色表示)Fig.2 HTPB/Al/AP/RDX four-component propellant model (The C/H/N/O/Al/Cl in the diagram are represented in gray/white/blue/red/silver/green respectively)
其中铝粉、AP、RDX均为颗粒物,它们之间使用HTPB进行填充。铝粉直径5nm,包含1nm厚度的氧化层;AP直径为20nm,RDX颗粒直径为5nm,两者均通过切割晶体结构获得。计算域大小为2nm×25nm×100nm,共计126165个原子。计算域的x和y方向设为周期性边界,z方向上设为反射边界。此外,推进剂模型下方5Å内原子均被固定住。为了模拟推进剂表面的燃烧过程,对模型的上表面区域(z>23.5nm)施加4000K高温热浴的正则系综,采用Nose-Hoover恒温器,阻尼因子设为20fs。其他区域采用微正则系综(NVE),初始温度设为500K,采用速度Verlet法对运动方程进行积分。分子动力学模拟计算使用LAMMPS软件[22],时间步长为0.2fs,共模拟40ps,在NVIDIA V100 GPU上运行约24h。
2 结果与讨论
2.1 机器学习势函数验证
用推进剂组分的第一性原理计算数据库对势函数进行测试,以评价势函数的准确性,结果见图3和表1。
数据库中的分子结构包括HTPB四组元推进剂(Al/RDX/AP/HTPB)的各个单质与两两界面构型。所有模型均使用CP2K在300~4000K下进行采样,每个温度区间各有1000个构型。
如图3所示,最终的四组元模型对于推进剂单质组分预测精度很高,对于不同组分组成的界面结构,误差略高于单质体系。表1中展示了每个体系的能量与受力误差。总体上,所有组分的MAE(平均绝对误差)均小于40meV,受力误差小于0.5eV/Å,这表明四组元势函数可较好地描述推进剂单质组分和界面上的化学反应过程。
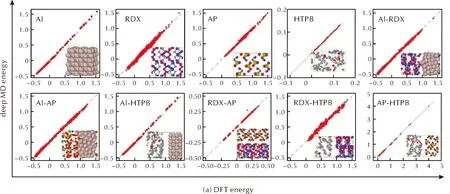
图3 单组分和两两界面的机器学习势函数的能量与受力精度测试Fig.3 Energy and force accuracy testing of machine learning potential for single-component and interfaces
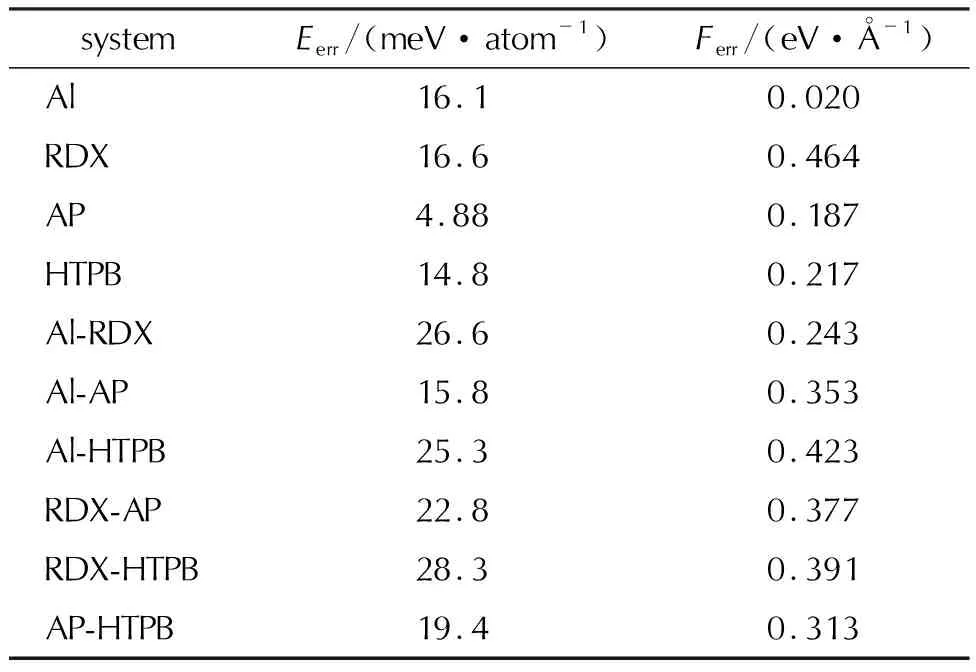
表1 机器学习势函数对推进剂单质和界面组分的能量与受力精度Table 1 Prediction accuracy of the machine learning potential function for energies and forces on single and interfacial components of solid propellants
2.2 推进剂复合模型燃烧结构演化
采用新开发的势函数模型对HTPB推进剂的燃烧过程进行全配方分子动力学模拟。对推进剂表面1.5nm区域内的原子施加4000K高温,其余部分温度为500K,以模拟推进剂燃面燃烧模型,燃烧过程如图4所示,初始时刻(t=2ps)推进剂表面的HTPB与RDX先发生分解反应,释放出气体小分子。随后在t=14ps时,HTPB进一步分解,气体小分子产物向燃面上方扩散,AP颗粒上方部分也发生分解。而铝粉反应较弱,在HTPB分解气体的推动下逐渐脱离燃面。在t=22ps时,AP上方形成锥形分解产物区,同时AP/HTPB界面上也观察到明显的分层现象。当t=34ps时,推进剂整体上密度显著降低,完全扩散至计算域中,生成气体分解产物。

图4 推进剂燃烧过程中结构演化Fig.4 Structural evolution during propellant combustion
进一步分析分子动力学模拟轨迹,可发现推进剂上方形成了扩散火焰结构。推进剂扩散火焰的局部放大图如图5所示,通过观察原子轨迹观察到此时RDX在高温区加速的条件下发生了快速分解,生成了大量NO2、H2O、N2O等产物。而AP颗粒则分解产生NO、HCl、O2等氧化性气体组分,这些组分可作为氧化剂与HTPB分解产生的碳氢小分子进行燃烧反应,主要反应区位于AP与HTPB的交界面处。铝粉虽然同样暴露在高温区,但是此时并没有直接参与燃烧反应,而是随着AP和HTPB分解产生的小分子气流向上运动。通过分子动力学模拟,可定量地对整个推进剂模型进行时间分辨的三维重建,进而获得推进剂燃烧的微观机理。
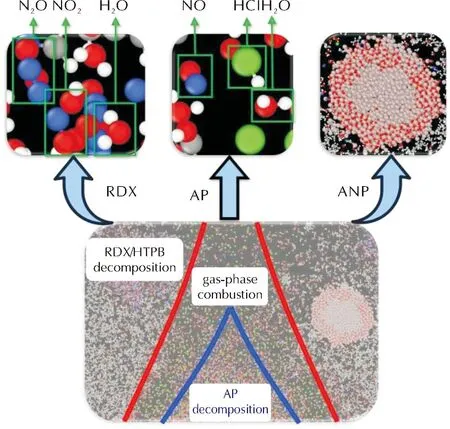
图5 推进剂扩散火焰微结构局部放大图(t=34ps)Fig.5 Local magnification of diffusion flame microstructure (t=34ps)
2.3 温度演化
此外,推进剂燃烧过程中的温度演化过程也可被完整地从分子模拟中重建出来。图6展示了不同时刻推进剂温度演化过程。
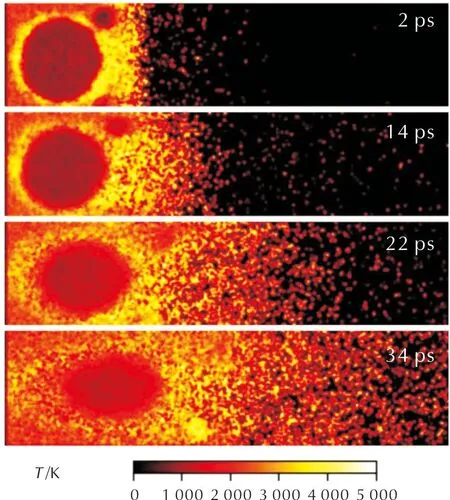
图6 推进剂燃烧过程中温度演化Fig.6 Temperature evolution during propellant combustion
初始时刻表面为4000K高温,其余部分为500K。随着热量的传递,推进剂上方温度快速升高。当t=2ps时,推进剂上方维持3000K左右的高温,RDX和AP颗粒由于快速分解反应的吸热效应,其所在区域温度显著降低。在AP周围与HTPB交界处,也观察到了显著的高温区,表明两者在界面上发生了小分子产物之间的放热反应。随着整体燃烧过程的推进,高温区逐渐从AP/HTPB交界处向两侧扩散,对AP/HTPB进行加热,促进两者的热分解过程。由于铝颗粒具有较好的热导率,其温度通过热传导,一直保持在较高的温度(约3000K)与扩散火焰中的温度基本保持一致。相对应的,AP颗粒内部的温度在模拟过程中缓慢的从约500K升高至约1000K。
2.4 推进剂燃烧中组分演化

由图7可知,除去部分热点区域,HClO4生成的位置均匀分布在整个AP颗粒内部,而并非是从AP/HTPB界面处向颗粒内部扩散。从时间演化上看,在初始时刻HClO4浓度较高(约1.5~2mol/cm3),随着反应的进行,HClO4浓度逐渐降至约0.5mol/cm3。这说明HClO4是整个燃烧过程中的一种中间产物,在后续反应中被快速消耗。
AP经过初始分解后,通过进一步的演化最终产生O2和Cl2,见图8,它们将作为四组元HTPB推进剂中最重要的两种氧化剂参与推进剂的燃烧过程。
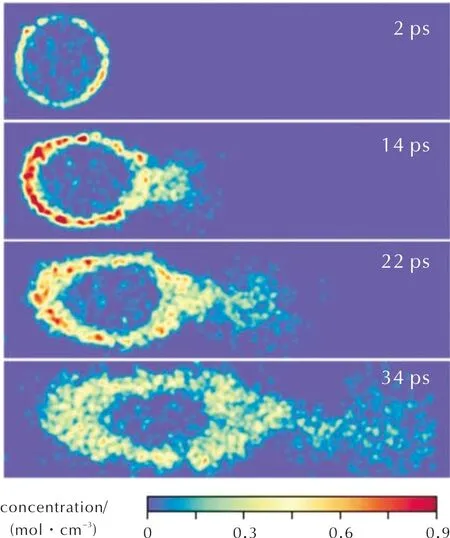
图8 O2/Cl2组分演化(图中浓度为两组分总浓度)Fig.8 Evolution of O2/Cl2 components(The concentration in the figure represents the total concentration of two components)
如图8所示,和HClO4相比,O2/Cl2的生成具有显著的空间依赖性,在AP/HTPB交界面处浓度较高,说明界面处AP与HTPB间的相互作用实际促进了O2/Cl2产生。这也为实验中观察到的HTPB对AP分解的促进作用提供了理论解释[7]。在14ps时,AP/HTPB界面处氧化剂浓度进一步升高,同时在推进剂上方产生了微弱的尖锥状气相产物区域,该部分O2和Cl2的浓度要比推进剂下方区域的浓度低2~3倍。这主要由于尖锥状区域的温度显著高于推进剂下方区域(图6),导致HTPB分解产物和O2/Cl2在该区域发生了快速的氧化反应,释放出了大量的能量。同时,这一尖锥状区域与图5所示的推进剂扩散火焰微结构一致,说明扩散火焰中主要发生的是HTPB分解的小分子碳氢化合物与O2/Cl2之间的气相反应。
在推进剂中也添加了少量的RDX颗粒。在此前研究中发现RDX热解的主要途径是通过N—N键均裂发生的,产生NO2和RDR分子[21]。随后NO2可通过进一步反应转化为NO。图9展示了NO/NO2的空间分布,可观察到在2ps时RDX的位置产生了大量的NO/NO2分子,同时在AP颗粒周围也有较少的分布。在推进剂组分的DSC实验中,前人观察到RDX的DSC峰温为242.7℃,低于AP的峰温296.7℃,这表明RDX更容易发生分解,与本研究的模拟结果一致。从浓度分布上看,RDX分解产生的NO/NO2分子浓度较高(大于0.4mol/cm3),而AP分解产生的NO/NO2分子浓度相对较低(小于0.15mol/cm3)。这些NO/NO2分子都将作为氧化剂与HTPB分解产物发生燃烧反应,最终转化为N2和H2O等物质。

图9 分解产物 NO/NO2分子的时空演化(图中浓度为两组分总浓度)Fig.9 The spatiotemporal evolution of NO/NO2 molecules(The concentration in the figure represents the total concentration of two components)
推进剂燃烧过程中,HTPB可分解产生一系列碳氢小分子,这些小分子可与AP分解产物形成扩散火焰,生成CO、CO2、H2O等燃烧产物。因为AP中不含C元素,所以通过CO/CO2的空间分布可具体确定扩散火焰发展的过程和具体形貌。图10展示了CO/CO2的时空演化过程,初始时刻(2ps)CO/CO2分子主要分布在AP/HTPB交界处,随着反应的进行,在推进剂AP颗粒上方逐渐形成了锥形的燃烧产物区。产物不断向上扩散,最终充满整个计算域。然而,在这个过程中,RDX颗粒附近并没有看到较多CO/CO2。如果比较RDX颗粒的空间位置(图9),能更清楚地发现RDX分解产物中没有CO/CO2。前期关于RDX的热分解过程研究发现RDX中CO/CO2的产生存在一定的滞后[21]。在2500K下观察到RDX先分解产生NO、NO2、HNO2等物质,在10ps左右后才观察到CO/CO2的生成。在推进剂燃烧中,推进剂表面的RDX先产生氮氧化物,RDX生成CO/CO2主要发生在气相反应区。
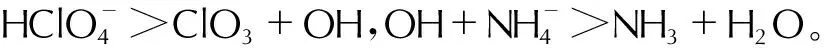

图10 CO/CO2组分演化(图中浓度为两组分总浓度)Fig.10 Evolution of CO/CO2 components(The concentration in the figure represents the total concentration of two components)
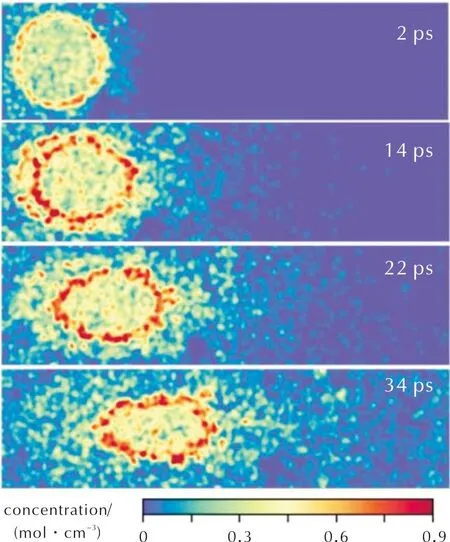
图11 H2O组分演化Fig.11 Evolution of H2O components
相对于HTPB/AP/RDX,铝粉与各个组分之间的反应相对较慢(图12)。随着燃烧的进行,可观察到铝粉颗粒快速向远离燃面的方向运动,但是反应程度有限。从组分分布来看,铝颗粒周围是3000K高温的H2O/CO2/O2/Cl2等氧化性气体,但是在本工作中,未发现这些氧化性气体与铝粉之间的反应。另外,从颗粒温度来看(图6),铝颗粒由于良好的热传导能力,在燃烧初期就已经达到了较高的温度,但是由于外层的氧化层存在,其反应活性被严重制约。即使本工作中为了加速潜在的铝粉与高温气体之间的反应,使用了1nm的超薄氧化层,仍然未发现铝粉参与反应的过程。在推进剂燃烧过程中,AP、HTPB等组分反应产生了大量气体,局部压力升高,推动铝颗粒离开推进剂表面,这一现象也在近期的推进剂燃烧实验的全息测量中得到了证实[8]。

图12 Al组分演化过程Fig.12 Evolution of Al components
3 结 论
(1)基于第一性原理计算的训练集,开发了一个通用势函数,可以开展HTPB/Al/AP/RDX推进剂关键组分的大规模分子动力学模拟,以研究推进剂的热解和燃烧。
(2)建立了HTPB四组元推进剂燃面模型,并开展分子动力学模拟研究推进剂的燃烧过程,实现了对温度、组分等信息的统计分析,发现了AP/HTPB间相互作用机制,AP分解产生的氧化剂在界面处与HTPB反应放热,进而促进了AP的分解过程。
(3)观察到了推进剂燃烧过程中铝粉的剥离过程,推进剂燃烧过程中产生大量气体,推动铝粉离开推进剂表面,而铝粉本身反应比较微弱。
(4)证明了分子动力学模拟能够在原子尺度上实现时间分辨的三维重建,进而获得推进剂燃烧的微观机理,为固体推进剂的理论研究提供了新的工具。
