钴配合物催化CO2氢化为甲醇的“氢质子和氢负离子”同时转移机理
2024-04-04闫秀丽赵雪乔石博郝勇静
闫秀丽 赵雪乔 石博 郝勇静
摘要:全球二氧化碳(CO2)过度排放,由此引发的温室效应日益严峻.将CO2选择性加氢为甲醇,可以将丰富、安全的碳源转化为有价值的化学品和燃料.文中采用密度泛函理论对半三明治结构钴配合物催化CO2加氢制甲醇的反应机理及新型催化剂的设计进行计算研究.计算结果表明,形成甲二醇分子时金属Co上的氢负离子和吡啶配体中的氢质子同时转移至甲酸分子上的过程是该反应的决速步,总反应能垒为122.2 kJ·mol-1(2→TS8,11).此外,通过不同的取代基取代酰胺氮原子和吡啶配体上的氢原子,设计了6种钴配合物,计算预测了它们催化CO2加氢到甲醇的活性.在所有新提出的钴配合物中,1a,1b,1c有潜力在温和条件下高效催化CO2加氢到甲醇,它们的总自由能垒分别为107.5,109.2和111.7 kJ·mol-1.
关键词:二氧化碳;催化加氢;密度泛函理论;反应机理;催化剂设计
中图分类号:O 643.36;TQ 223.121 文献标志码:A 文章编号:1001-988Ⅹ(2024)02-0058-07
Simultaneous transfer mechanism of“a hydroxyl proton and hydride”for the hydrogenation of CO2to methanol catalyzed by cobalt complexes
YAN Xiu-li ZHAO Xue-qiao ,SHI Bo3,HAO Yong-jing1
Abstract:The greenhouse effect caused by excessive global carbon dioxide(CO2) emissions is becoming more and more serious.Selective hydrogenation of CO2 to methanol can convert abundant and safe carbon sources into valuable chemicals and fuels.A computational study using density functional theory is conducted to investigate the reaction mechanism of CO2 hydrogenation to methanol catalyzed by a half-sandwich cobalt complex,as well as the design of new catalysts.The calculated results show that the formation of methanediol via the simultaneous transfer of a hydroxyl proton in the pyridine ligand and the hydride on the metal Co to the formic acid is the rate-determining step with a total barrier of 122.2 kJ·mol-1(2→TS8,11).Furthermore,six cobalt complexes are designed by replacing the hydrogen atom on the amide nitrogen atom and pyridine ligand with different substituents.Among all newly proposed cobalt complexes,1a,1b,and 1c are the promising catalyst candidates for hydrogenation of CO2 to methanol with low total free energy barriers of 107.5,109.2,and 111.7 kJ·mol-1,respectively.
Key words:carbon dioxide;catalytic hydrogenation;density functional theory;reaction mechanism;catalyst design
根據英国BP石油公司统计的2022年世界能源消耗结果,全球对于煤炭、石油、天然气等的能源消耗总量依旧不断增加,化石燃料仍为目前能源的主要来源[1].过度使用化石能源也引起了严重的环境问题,如二氧化碳(CO2)过度排放导致全球气候变化和温室效应[2].据统计,在1960~2023年,大气中CO2的浓度已由工业化前的2.8×10-4上升至4.19×10-4,如果全球能源需求继续增长并且主要通过化石燃料来满足,那么到本世纪末,大气中的CO2浓度预计将超过9.0×10-4,这可能带来更加强烈的气候变化.在这种背景下,氢(H2)作为一种清洁的替代能源引起了科学家们越来越多的关注,目前主要通过化石燃料的燃烧以及电解水制氢[3],但温室气体的排放和较高的生产成本使得发展经济、可持续的氢能源仍处在瓶颈期.
CO2作为自然界中丰富、安全、廉价的C1资源,在循环利用的诸多方式中,可以实现高附加值化学品和燃料的合成,对于实现人类社会的可持续发展具有重要的意义和广泛的应用前景.CO2加氢制甲醇反应(CO2+3H2→CH3OH+H2O)是实现CO2转化利用的一种理想方式.这是因为甲醇既可以作为代用燃料,又具有较高的氢含量(12.6%)和方便运输的优点[4-5].
近年来,科研工作者对催化CO2转化的反应进行了大量研究[4,6-11].Schneidewind等[12]报道了一种均相非贵金属Co配合物,其催化CO2直接加氢到甲醇的TON仅为78.Kar等[13]报道了Mn-PNP 配合物均相催化CO2加氢到甲醇,反应的最大TON仅为36.Schieweck等[11]报道了三齿磷配体螯合的Ru配合物,在乙醇溶剂中催化CO2直接加氢到甲醇的TON最高可达2100.Kanega等[14]报道了在温和反应条件下(30 ℃,5 MPa(TON 2.0)和70 ℃,0.5 MPa(TON 3.0)),使用多核过渡金属Ir配合物加氢制备甲醇的新方法.Kanega等[10]报道了一系列Ir配合物催化CO2加氢和甲酸脱氢的反应,其中含酰胺配体的铱配合物Cp*Ir(L12)(H2O)HSO4(L12=6-hydroxy-N-phenylpicolinamidate)在反应初始1 h内的TOF为198 h-1.尽管关于均相催化CO2加氢到甲醇的报道已经取得了一定的进展,但是大多数实验报道的催化体系都存在贵金属以及对空气和水敏感的磷配体,且反应条件严苛,催化效率低下.因此,发展可催化CO2加氢到甲醇的低成本、高效率以及不含磷配体的过渡金属催化剂依然是亟待解决的重要问题.
受Kanega等[14]报道的半三明治结构铱配合物相关催化反应机理的启发,文中计算了非贵金属钴配合物[Cp*Co(L)OH2]+(1,L=2-hydroxy-N-picolinamidate)催化CO2加氢到甲醇的详细反应机理.在明确反应机理的基础上,构建了6种钴配合物,计算了其催化CO2加氢到甲醇的反应中潜在的关键中间体和过渡态之间的相对吉布斯自由能,从而提出了可以高效催化该反应的非贵金属催化剂结构.
1 研究方法
文中所有的 DFT 计算均使用M06 泛函[15],并在Gaussian 09程序包[16]中完成.为了提高计算效率,在保证计算精度的前提下,对所有C,H原子采用6-31G(d,p)基组[17],N,O原子采用6-31+G(d)[18]基组,Co原子采用包含相对论效应修正的Stuttgart系列赝势基组ECP10MDF[19-20].所有构型优化都考虑了溶剂效应的影响,采用SMD溶剂模型[21](极性部分使用IEFPCM方法[22]进行计算).对体系中所有结构均进行溶剂校正,文中以水作为溶剂.质子在水溶剂中的自由能Gsol(H+)使用实验值-1 112.5 kJ·mol-1[23].在T为298.15 K和1 atm大氣压条件下进行热力学能量修正.采用超精细积分格点(99,590)进行数值积分计算.同时,对所有优化后的几何构型进行频率计算,结果表明所有中间体均无虚频,过渡态有且只有一个虚频.所有过渡态均通过内禀反应坐标法(IRC)确认能够连接正确的中间体.3D分子结构图由JIMP2软件[24]绘制.
2 结果与讨论
2.1 CO2加氢到甲酸
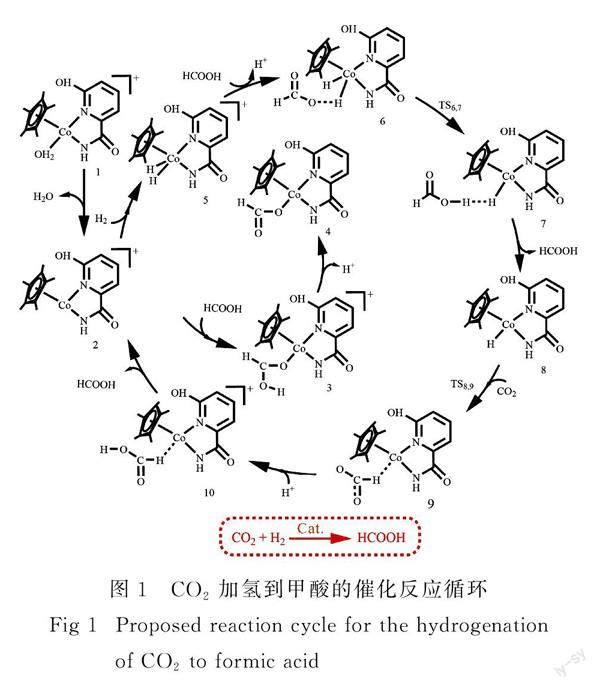
图1为1催化CO2加氢到甲酸的催化反应循环(Cycle 1).
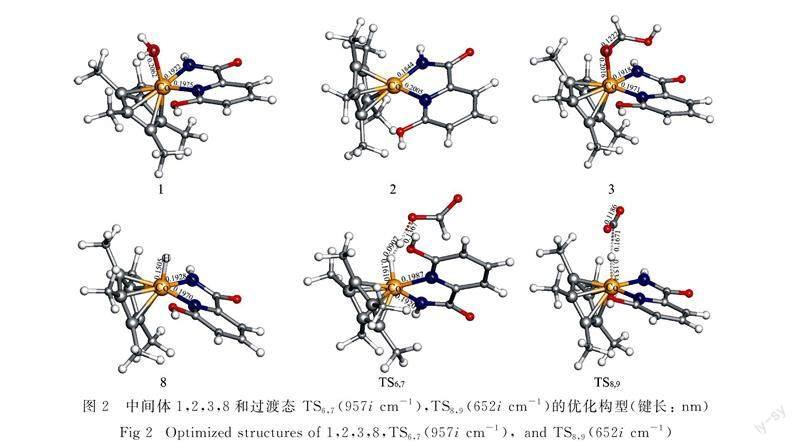
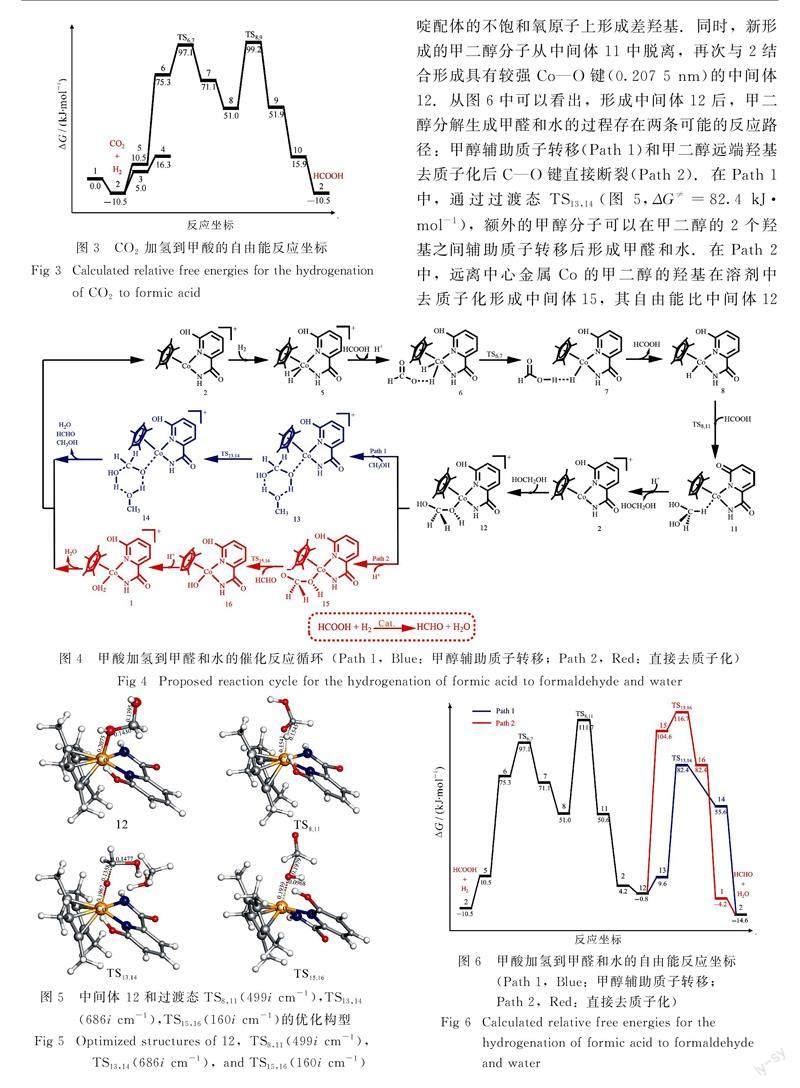
在反应的开始,催化剂1的中心金属Co原子失去配位的H2O分子,经过10.5 kJ·mol-1的自由能下降过程,形成中间体2.随后,H2分子占据中间体2的空位后形成自由能为10.5 kJ·mol-1的中间体5.之后中间体5中H2上的氢质子通过过渡态TS6,7(图2,ΔG≠=97.1 kJ·mol-1)转移到溶液中甲酸根阴离子的氧原子上形成甲酸.新形成的甲酸分子从中间体7中脱去后形成金属氢化物8.接着,一个CO2分子与8反应,通过过渡态TS8,9(图2,ΔG≠=99.2 kJ·mol-1)夺取中心金属Co上的氢负离子到其不饱和的碳原子上,得到甲酸根阴离子.中间体9中的甲酸根阴离子从溶剂中得到一个氢质子,然后从10中以甲酸分子的形式离去.从自由能反应坐标图可以看出(图3),中间体2和过渡态TS8,9是CO2加氢到甲酸过程的决速态,能量跨度为109.7 kJ·mol-1(2→TS8,9).由于催化剂1并没有参与该催化循环,因此可以被认为是该反应的预催化剂.除了H2,甲酸也可以占据中间体2 的空位,形成具有较强Co—O键(0.206 1 nm)的稳定中间体3.
2.2 甲酸加氢到甲醛和水
图4为1催化甲酸加氢到甲醛和水的催化反应循环(Cycle 2).
甲酸加氢生成甲醛和水的反应由两个催化循环构成:甲酸加氢生成甲二醇以及甲二醇分解生成甲醛和水.在该循环的开始与Cycle 1类似,都是形成中间体8.然后甲酸分子与8结合,通过过渡态TS8,11(图5, ΔG≠=111.7 kJ·mol-1)同时夺取中心金属Co上的氢负离子与吡啶配体中的氢质子到其不饱和碳原子和氧原子上形成中间
体11. 随后, 溶剂向中间体11转移一个质子到吡啶配体的不饱和氧原子上形成差羟基.同时,新形成的甲二醇分子从中间体11中脱离,再次与2结合形成具有较强Co—O键(0.207 5 nm)的中间体12.从图6中可以看出,形成中间体12后,甲二醇分解生成甲醛和水的过程存在两条可能的反应路径:甲醇辅助质子转移(Path 1)和甲二醇远端羟基去质子化后C—O键直接断裂(Path 2).在Path 1中,通过过渡态TS13,14(图 5,ΔG≠=82.4 kJ·mol-1),额外的甲醇分子可以在甲二醇的2个羟基之间辅助质子转移后形成甲醛和水.在Path 2中,远离中心金属Co的甲二醇的羟基在溶剂中质子化形成中间体15,其自由能比中间体12高105.4 kJ·mol-1.然后通过过渡态TS15,16(图5, ΔG≠=116.7 kJ·mol-1),经过 C—O 键直接断裂后生成产物甲醛分子,该过程的自由能仅比中间体15高12.1 kJ·mol-1.最后,中间体16中的羟基从溶剂中得到一个质子,以水分子的形式从1中脱离再次生成中间体2.比较图4中的两条反应路径,从自由能反应坐标图(图6)可以看出,Path 1中甲醇辅助质子转移(TS13,14)的自由能垒比Path 2中C—O键直接断裂(TS15,16)要低34.3 kJ·mol-1.通过分析该循环的自由能反应坐标图(图6),中间体2和TS8,11是甲酸加氢到甲醛和水反应过程的决速态,能量跨度为122.2 kJ·mol-1(2→TS8,11).
2.3 甲醛加氢到甲醇
图7为1催化甲醛加氢到甲醇的催化反应循环(Cycle 3).
与甲酸加氢反应类似,Cycle 3 的开始也是先通过过渡态TS6,7形成中间体 8.随后甲醛分子可通过TS17,18(图8, ΔG≠=73.2 kJ·mol-1)夺取金属 Co上的氢负离子到其不饱和碳原子上,该过程跨越的自由能垒低于TS6,7(图9).之后中间体18从溶剂中得到质子后以甲醇分子的形式脱去,再次生成中间体2,该过程自由能下降61.0 kJ·mol-1.甲醇分子也可以再次与2结合,形成稳定的中间体20.该中间体由于具有较强的Co—O 键(0.204 7 nm),因此其自由能比中间体2稳定72.3 kJ·mol-1.除了形成20,甲醛分子也可以占据中间体2的空位,形成自由能比分离的中间体2和甲醛高1.3 kJ·mol-1的中间体19.
通过比较上述3个催化循环的自由能反应坐标,整个催化循环的决速步为形成甲二醇分子时金属Co上的氢负离子和吡啶配体中的氢质子同时转移至甲酸分子上的步骤,总反应能垒为122.2 kJ·mol-1(2→TS8,11).
2.4 计算设计新型的催化剂
为了找到催化活性较高的潜在过渡金属Co催化剂,使用不同取代基取代酰胺氮原子和吡啶配体上的氢原子,并设计了6种钴配合物(图10).图10给出了新设计的钴配合物催化CO2加氢反应中决速步骤之间的相对吉布斯自由能.可以看到,1e和1f具有相对较高的反应能垒,分别为126.8和128.9 kJ·mol-1,因此它们在水溶液中催化这类反应并不太容易.而催化剂1a,1b,1c具有相对较低的反应能垒,分别为107.5,109.2,111.7 kJ·mol-1,较低的能垒表明,在温和的条件下可以有效地催化CO2加氢到甲醇.
为了深入了解催化活性和电子结构之间的关系,对1,8 和 TS8,11进行了自然键轨道(NBO)分析.从表1可知,1a-1f中金属 Co 上的NPA电荷差距不大,但是 1a-1c中与 Co 相连的吡啶配体N原子的NPA电荷比1d-1f更负.对于金属氢化物(8a-8f), 8a-8f中金属 Co 上的NPA电荷差距不大,8a-8c中与Co相连的吡啶配体N原子的NPA电荷同样比8d-8f更负,而H原子上的正电荷比8d-8f更少.因此,由于电荷分布的不同使得催化剂1a,1b,1c催化反应的自由能垒相对较低.
3 结论
对半三明治结构的钴配合物催化CO2加氢反应进行了详细的计算研究并提出了“氢质子和氢负离子”同时转移的机理.整个催化循环的决速步为形成甲二醇反应中Co上的氢负离子和吡啶配体中的氢质子同时转移至甲酸分子上的步骤,总反应能垒为122.2 kJ·mol-1(2→TS8,11).使用不同的取代基取代酰胺氮原子和吡啶配体上的氢原子,构建并预测了6种半三明治结构的钴配合物.通过比较决速步的相对吉布斯自由能,研究了其催化CO2加氢反应的活性.计算结果表明,催化剂1a,1b,1c具有相对较低的反应能垒,分别为107.5,109.2, 111.7 kJ·mol-1.
这样较低的能垒表明,在温和的条件下可以有效地催化CO2加氢到甲醇.在此项工作中,通过对非贵金属钴配合物催化CO2加氢的反应机理研究和对不同取代基影响反应能垒的分析,为进一步设计可用于催化CO2加氢到甲醇的非贵金属催化剂提供了思路.
参考文献:
[1]POORMOHAMMADIAN S J,BAHADORAN F,VAKILI-NEZHAAD G R.Recent progress in homogeneous hydrogenation of carbon dioxide to methanol[J].Rev Chem Eng,2023,39(5):783.
[2]ONISHI N,HIMEDA Y.Homogeneous catalysts for CO2 hydrogenation to methanol and methanol dehydrogenation to hydrogen generation[J].Coord Chem Rev,2022,472:214767.
[3]SCHNEIDEWIND J,ARGELLO C M A,JUNGE H,et al.Two-photon,visible light water splitting at a molecular ruthenium complex[J].Energy Environ Sci,2021,14(8):4427.
[4]NAVARRO-JAN S,VIRGINIE M,BONIN J,et al.Highlights and challenges in the selective reduction of carbon dioxide to methanol[J].Nat Rev Chem,2021,5(8):564.
[5]NITOPI S,BERTHEUSSEN E,SCOTT S B,et al.Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte[J].Chem Rev,2019,119(12):7610.
[6]DU X L,JIANG Z,SU D S,et al.Research progress on the indirect hydrogenation of carbon dioxide to methanol[J].Chem Sus Chem,2016,9(4):322.
[7]田海峰,廖建康,查飛,等.丙烷与二氧化碳耦合制丙烯的热力学模拟研究[J].西北师范大学学报(自然科学版),2019,55(2):94.
[8]WESSELBAUM S,VOM S T,KLANKERMAYER J,et al.Hydrogenation of carbon dioxide to,methanol by using a homogeneous ruthenium-phosphine catalyst[J].Angew Chem Int Ed,2012,51(30):7499.
[9]BERNSKOETTER W H,HAZARI N.Reversible hydrogenation of carbon dioxide to formic acid and methanol:Lewis acid enhancement of base metal catalysts[J].Acc Chem Res,2017,50(4):1049.
[10]KANEGA R,ERTEM M Z,ONISHI N,et al.CO2 hydrogenation and formic acid dehydrogenation using ir catalysts with amide-based ligands[J].Organometallics,2020,39(9):1519.
[11]SCHIEWECK B G,JRLING-WILL P,KLANKERMAYER J.Structurally versatile ligand system for the ruthenium catalyzed one-pot hydrogenation of CO2 to methanol[J].ACS Catalysis,2020,10(6):3890.
[12]SCHNEIDEWIND J,ADAM R,BAUMANN W,et al.Low-temperature hydrogenation of carbon dioxide to methanol with a homogeneous cobalt catalyst[J].Angew Chem Int Ed,2017,56(7):1890.
[13]KAR S,GOEPPERT A,KOTHANDARAMAN J,et al.Manganese-catalyzed sequential hydrogenation of CO2 to methanol via formamide[J].ACS Catalysis,2017,7(9):6347.
[14]KANEGA R,ONISHI N,TANAKA S,et al.Catalytic hydrogenation of CO2 to methanol using multinuclear iridium complexes in a gas-solid phase reaction[J].J Am Chem Soc,2021,143(3):1570.
[15]ZHAO Y,TRUHLAR D G.The M06 suite of density functionals for main group thermochemistry,thermochemical kinetics,noncovalent interactions,excited states,and transition elements:two new functionals and systematic testing of four M06-class functionals and 12 other functionals[J].Theor Chem Acc,2008,120(1-3):215.
[16]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09,Revision E.01[Z].Wallingford CT:Gaussian,Inc.,2010.
[17]PERDEW J P,BURKE K,ERNZERHOF M.Generalized gradient approximation made simple[J].Phys Rev Lett,1996,77(18):3865.
[18]FRANCL M M,PIETRO W J,HEHRE W J,et al.Self-consistent molecular orbital methodsⅩⅫ.A polarization-type basis set for second-row elements[J].J Chem Phys,1982,77(7):3654.
[19]ANDRAE D,HUSSERMANN U,DOLG M,et al.Energy-adjusted Ab initio pseudopotentials for the second and third row transition elements[J].Theoretica Chimica Acta,1990,77(2):123.
[20]MARTIN J M L,SUNDERMANN A.Correlation consistent valence basis sets for use with the stuttgart-dresden-bonn relativistic effective core potentials:the atoms Ga-Kr and In-Xe[J].J Chem Phys,2001,114(8):3408.
[21]MARENICH A V,CRAMER C J and TRUHLAR D G.Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions[J].J Phys Chem B,2009,113(18):6378.
[22]TOMASI J,MENNUCCI B,CAMMI R.Quantum mechanical continuum solvation models[J].Chem Rev,2005,105(8):2999.
[23]CAMAIONI D M,SCHWERDTFEGER C A.Comment on“accurate experimental values for the free energies of hydration of H+,OH-,and H3O+”[J].J Phys Chem A,2005,109(47):10795.
[24]MANSON J,WEBSTER C E,HALL M B.JIMP2,Version 0.091[Z].College Station,TX:Texas A&M University,2006.
(责任编辑 陆泉芳)
收稿日期:2023-09-28;修改稿收到日期:2023-12-23
基金項目:河北省自然科学基金资助项目(B2021402012; E2021402017)
作者简介:闫秀丽 (1989—),女,河北邯郸人,讲师,博士.主要研究方向为理论有机催化反应.E-mail:yanxiuli@hebeu.edu.cn
*通信联系人,女,副教授,博士.主要研究方向为催化化学.E-mail:haoyj@hebeu.edu.cn
