萘查耳酮类PTP1B 抑制剂的合成与活性研究
2024-03-25施晓文郭乔月廖远峰邓先清
施晓文,郭乔月,廖远峰,邓先清
(1.井冈山大学医学部,江西,吉安 343009;2.吉安市科技创新发展中心,江西,吉安 343000)
蛋白酪氨酸磷酸酯酶(PTP)是一类功能性酶,调节并涉及众多生物学和病理事件中的去磷酸化过程[1-3]。在人类中存在100 多种PTP,可以在各种信号转导途径中作为负向或正向调节因子发挥作用[4]。PTP 活性的异常与许多人类疾病相关,包括癌症、糖尿病、肥胖症和自身免疫低等疾病[5]。PTP1B 作为PTP 超家族的一员,通过作为胰岛素和瘦素受体所介导信号途径的重要负调节因子,在调节体重、葡萄糖稳态和能量消耗方面发挥关键作用[6-7]。PTP1B 可以通过去磷酸化胰岛素受体及其下游的一些信号来转导蛋白,并降低胰岛素信号[8-9]。
研究发现,PTP1B 缺陷的小鼠基础代谢率增高,胰岛素敏感性增强,在高脂饮食下也不容易肥胖[10-11]。而且进一步研究发现,PTP1B 缺陷造成的胰岛素敏感性增强具有组织特异性,仅限于骨骼肌,脂肪组织的胰岛素敏感性不会变化。也就是说,抑制PTP1B 并不会促进脂肪组织摄取葡萄糖而造成脂肪堆积。因此,PTP1B 抑制剂被认为是治疗糖尿病和肥胖症的新希望。除了通过介导胰岛素信号通路来治疗代谢疾病与肥胖症外,PTP1B 还在树突细胞的成熟和迁移、T 细胞的活化中起了关键的调控作用。因此靶向PTP1B 还可以用来激活免疫而治疗癌症[12]。最近Tony Tiganis 等人发现PTP1B 可能成为继PD-1/PD-L1 之后,新一代的免疫检查点抑制剂。其研究发现PTP1B 可以抑制T 细胞中的STAT5 信号,从而抑制CD8+T细胞的增殖和功能。而PTP1B 的缺陷可以促进T细胞的扩增和激活,增强其抗肿瘤活性。在小鼠实验中,PTP1B 抑制剂MSⅠ-1436 增加了肿瘤中免疫细胞的浸润而抑制了肿瘤生长,并提高PD-1 抑制剂和CAR-T 的疗效[13]。
基于高通量筛选和基于结构的药物设计,各制药公司在过去几十年间已经陆续开发了各种PTP1B 抑制剂,比如二氟亚甲基磷酸酯(DFMP)、草酸氨基苯甲酸(OBA)和磷酸化酪氨酸模拟物等[7]。然而,其有限的细胞膜渗透性和选择性阻碍了它们进入临床阶段[14]。这些化合物中高电荷的药效团(如磷酸、羧酸、磺酸)是其低生物利用度的主要原因。因此,寻找具有高PTP1B 抑制活性的非酸性配体是一项值得研究的工作。
查尔酮是一类可以从天然的可食用植物中分离出来的黄色物质,被认为是黄酮类和异黄酮类的前体。作为黄酮类家族的一个子类,查尔酮衍生物已被报道具有多种生物活性,包括抗癌[15]、抗真菌[16]、抗炎[17]、抗微生物[18]、抗氧化[19]、抗抑郁等[20]。并且查尔酮结构毒性低,代谢较稳定,具有良好的药物潜力。因此,查尔酮是一类具有重要治疗潜力的化合物。
Yoon 等人从天然产物及其衍生物中已鉴定出多种具有PTP1B 抑制剂活性的查尔酮类物质(如图1 所示)[21]。这类查尔酮通过抑制PTP1B 来改善葡萄糖吸收,发挥与二甲双胍等临床抗糖尿病药物相类似的降血糖作用。它们的共同特点是具有羟基查尔酮骨架,在PTP1B 的抑制中发挥了关键作用[22]。天然产物通常由复杂的结构组成,但通过对它们进行简化或者非药效团的修饰,并不会改变其生物活性。从天然产物可卡因的结构简化中发现普鲁卡因盐酸盐就是一个经典例子。
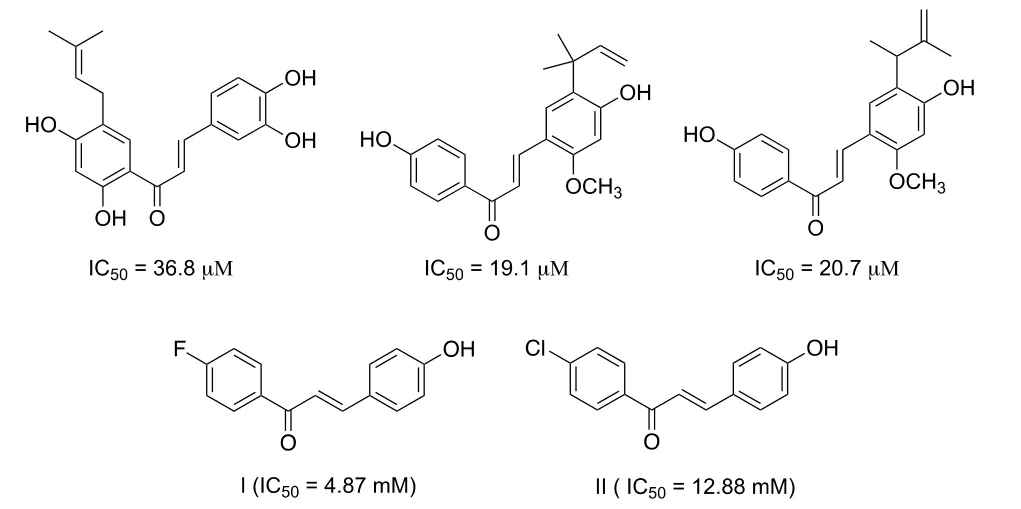
图1 具有PTP1B 抑制活性的羟基查尔酮衍生物Fig.1 The chalcone derivatives identified as PTP1B inhibitors
基于该思路,在前期工作中,本课题组制备了一系列简单的羟基查尔酮及其环化衍生物,并评估了它们对PTP1B 酶的抑制活性[23]。结果发现大部分化合物具有较好的PTP1B 抑制活性,其中两个化合物(图1,Ⅰ和ⅠⅠ)对PTP1B 的抑制活性ⅠC50分别为4.87、12.88 μM。将化合物Ⅰ与PTP1B蛋白进行了分子对接(见图2),结果显示结构中酚羟基与ASP181 氨基酸残基产生氢键结合,是其与PTP1B 结合的关键基团,并且发现查尔酮中羟基所在苯环与蛋白存在较大空隙,增大该部位的体积,可能提高与PTP1B 酶的结合力,从而提高PTP1B 抑制活性。因此,为了进一步丰富查尔酮衍生物的PTP1B 抑制活性构效关系,发现活性更好的PTP1B 抑制剂,本实验设计并合成了一系列萘查耳酮衍生物,评价了化合物的PTP1B 抑制活性,希望通过增加化合物与PTP1B 蛋白的相互作用力,从而提高PTP1B 抑制活性。
1 材料与方法
1.1 仪器和试剂
所有试剂和溶剂均购自阿拉丁试剂(中国上海)或国药集团化学试剂有限公司(中国上海)。熔点测定采用开放式毛细管,未经修正。反应过程采用薄层色谱监测(德国默克公司)。核磁共振波谱采用AV-300 光谱仪(瑞士布鲁克)在300 MHz 下进行核磁共振氢谱测定,使用DMSO-d6和CDCl3作为溶剂,四甲基硅烷作为内标准。质谱测定采用HP1100LC 质谱仪(美国安捷伦技术公司)。
1.2 化合物的合成
1.2.1 化合物1a-1g 的合成方法
以化合物1a 为例:将6 -羟基-2 -萘甲醛(3.44 g,20 mmol)和苯乙酮(2.40 g,20 mmol)加入含20 mL 无水乙醇的圆底烧瓶中,搅拌下缓慢加入20 mL 25% NaOH 溶液。混合物在室温继续搅拌48 h,TLC 监控反应,待反应完全后,用10%稀盐酸酸化至pH 值为3。黄色析出物过滤,滤饼用乙醇重结晶得到化合物1a。相同方法下,用其他取代苯乙酮和6-羟基-2-萘甲醛制备得到化合物1b-1g。
1.2.2 化合物2a-2g 的合成方法
以化合物2a 为例:将羟基萘查尔酮(1.12 g,5 mmol)和乙酰肼(370 mg,5 mmol)混合于10 mL乙醇中,在氮气保护下回流3 ~8 h。TLC 监控反应,反应完全后,加入20 mL 水,在冰水浴下迅速搅拌。放置后过滤,滤饼用乙醇重结晶得到化合物2a。相同方法下,用1b-1g 和乙酰肼制备得到化合物2b-2g。
1.3 目标化合物波谱数据
(E)-3-(6-羟基萘-2-基)-1-苯基丙-2-烯-1-酮(1a).Yield 76%,m.p.200-202 ℃.1H-NMR(DMSOd6+ CDCl3, 300 MHz)δ: 7.21-7.29 (m, 4H, Ph-H),7.83 (d, 1H,J= 15.5 Hz, CH=C), 7.80-7.90 (m, 4H,Ph-H),7.94(d,1H,J=15.5 Hz,CH=C),7.99-8.11(m,3H,Ph-H),9.51(br.s,1H,OH).MSm/z275(M+1)。
(E)-1-(4-氟苯基)-3-(6-羟基萘-2-基)丙-2-烯-1-酮(1b).Yield 69%, m.p.230-232 ℃.1H-NMR(DMSO-d6+ CDCl3, 300 MHz)δ: 7.16-7.25 (m, 4H,Ph-H),7.60(d,1H,J=15.6 Hz,CH=C),7.73-7.79(m,3H, Ph-H), 7.94 (d, 1H,J= 15.6 Hz, CH=C), 7.94 (s,1H, Ph-H), 8.09-8.14 (m, 2H, Ph-H), 9.52 (br.s, 1H,OH).MSm/z293(M+1)。
(E)-1-(4-氯苯基)-3-(6-羟基萘-2-基)丙-2-烯-1-酮(1c).Yield 77%, m.p.229-231 ℃.1H-NMR(DMSO-d6+CDCl3,300 MHz)δ:7.16(d,2H,J=7.5 Hz, Ph-H), 7.46 (d, 1H,J= 15.7 Hz, CH=C), 7.51 (s,1H, Ph-H), 7.56 (d, 1H,J= 15.7 Hz, CH=C), 7.65-7.77(m,3H,Ph-H),7.90-8.02(m,4H,Ph-H),9.47(br.s,1H,OH).MSm/z309(M+1)。
(E)-1-(2,4-二氯苯基)-3-(6-羟基萘-2-基)丙-2-烯-1-酮(1d).Yield 65%, m.p.218-220 ℃.1HNMR(DMSO-d6+CDCl3,300 MHz)δ:7.15-7.17(m,2H, Ph-H), 7.38-7.51 (m, 4H, Ph-H, CH=C), 7.56 (d,1H,J= 16.0 Hz, CH=C), 7.62-7.73 (m, 3H, Ph-H),7.86 (s, 1H, Ph-H), 9.53 (s, 1H, OH).MSm/z344(M+1)。
(E)-1-(4-溴苯基)-3-(6-羟基萘-2-基)丙-2-烯-1-酮(1e).Yield 74%, m.p.231-233 ℃.1H-NMR(DMSO-d6+CDCl3,300 MHz)δ:7.17(d,2H,J=7.4 Hz, Ph-H), 7.45 (s, 1H, Ph-H), 7.56 (d, 1H,J= 15.6 Hz, CH=C), 7.65-7.77 (m, 5H, Ph-H, CH=C), 7.91-7.95(m,3H,Ph-H),9.50(br.s,1H,OH).MSm/z353(M+1)。
(E)-1-(4-甲基苯基)-3-(6-羟基萘-2-基)丙-2-烯-1-酮(1f).Yield 74 %, m.p.210-211 ℃.1HNMR (DMSO-d6+ CDCl3, 300 MHz)δ: 2.45 (s, 3H,CH3),7.17(s,1H,Ph-H),7.33(d,2H,J=8.1 Hz,Ph-H), 7.52 (s, 1H, Ph-H), 7.62 (d, 1H,J= 15.5 Hz,CH=C),7.71(d,1H,J=15.5 Hz,CH=C),7.75(d,2H,J= 8.1 Hz, Ph-H), 7.88-7.98 (m, 4H, Ph-H), 9.50(br.s,1H,OH).MSm/z289(M+1)。
(E)-1-(4-甲氧基苯基)-3-(6-羟基萘-2-基)丙-2-烯-1-酮(1g).Yield 79%, m.p.200-203 ℃.1HNMR (DMSO-d6+ CDCl3, 300 MHz)δ: 3.84 (s, 3H,OCH3), 6.94 (d, 2H,J= 8.8 Hz, Ph-H), 7.12 (s, 1H,Ph-H), 7.32 (s, 1H, Ph-H), 7.55 (d, 1H,J= 15.6 Hz,CH=C), 7.61-7.69 (m, 3H, Ph-H), 7.85 (d, 1H,J=15.6 Hz, CH=C), 7.86 (s, 1H, Ph-H), 8.01 (d, 2H,J=8.8 Hz, Ph-H), 9.30 (br.s, 1H, OH).MSm/z305(M+1)。
1-(5-(6-羟基萘-2-基)-3-苯基-4,5-二氢-1H-吡唑-1-基)乙-1-酮(2a).Yield68%,m.p.239-241 ℃.1H NMR (DMSO-d6, 300 MHz)δ: 2.33 (s, 3H,COCH3), 3.22 (dd, 1H,J1= 4.7 Hz,J2= 18.2 Hz,CH2-Ha), 3.90 (dd, 1H,J1=11.8 Hz,J2= 18.2 Hz,CH2-Hb),5.64(dd,1H,J1=4.7 Hz,J2=11.8 Hz,CH),7.05-7.83 (m, 11H, Ar-H), 9.71 (s, 1H, OH).MSm/z331(M+1)。
1-(3-(4-氟苯基)-5-(6-羟基萘-2-基)-4,5-二氢-1H-吡唑-1-基)乙-1-酮(2b).Yield 64%,m.p.253-255 ℃.1H NMR(DMSO-d6,300 MHz)δ:2.30(s,3H,COCH3),3.20(dd,1H,J1=4.8 Hz,J2=18.6 Hz,CH2-Ha), 3.88 (dd, 1H,J1=11.6 Hz,J2= 18.5 Hz,CH2-Hb),5.62(dd,1H,J1=4.8 Hz,J2=11.6 Hz,CH),7.03-7.87 (m, 9H, Ar-H), 9.71 (s, 10H, OH).MSm/z349(M+1).
1-(3-(4-氯苯基)-5-(6-羟基萘-2-基)-4,5-二氢-1H-吡唑-1-基)乙-1-酮(2c).Yield 67%, m.p.279-281 ℃.1H NMR(DMSO-d6,300 MHz)δ:2.30(s,3H,COCH3),3.20(dd,1H,J1=4.8 Hz,J2=18.4 Hz,CH2-Ha), 3.88 (dd, 1H,J1=11.9 Hz,J2= 18.4 Hz,CH2-Hb),5.62(dd,1H,J1=4.8 Hz,J2=11.9 Hz,CH),7.03-7.82 (m, 10H, Ar-H), 9.72 (s, 1H, OH).MSm/z365(M+1)。
1-(3-(2,4-二氯苯基)-5-(6-羟基萘-2-基)-4,5-二氢-1H-吡唑-1-基)乙-1-酮(2d).Yield 52 %,m.p.230-232℃.1H NMR (DMSO-d6, 300 MHz)δ:2.28 (s, 3H, COCH3), 3.26 (dd, 1H,J1= 4.6 Hz,J2=18.5 Hz, CH2-Ha), 4.00 (dd, 1H,J1=11.8 Hz,J2=18.5 Hz,CH2-Hb),5.61(dd,1H,J1=4.6 Hz,J2=11.8 Hz, CH), 7.04-7.82 (m, 9H, Ar-H), 9.73 (s, 1H, OH).MSm/z399(M+1)。
1-(3-(4-溴苯基)-5-(6-羟基萘-2-基)-4,5-二氢-1H-吡唑-1-基)乙-1-酮(2e).Yield 69%, m.p.292-294℃.1H NMR(DMSO-d6,300 MHz)δ:2.30(s,3H,COCH3),3.20(dd,1H,J1=4.4 Hz,J2=18.5 Hz,CH2-Ha), 3.88 (dd, 1H,J1=12.2 Hz,J2= 18.5 Hz,CH2-Hb),5.62(dd,1H,J1=4.4 Hz,J2=12.2 Hz,CH),7.03-7.75 (m, 10H, Ar-H), 9.72 (s, 1H, OH).MSm/z409(M+1)。
1-(5-(6-羟基萘-2-基)-3-(对甲苯基)-4,5-二氢-1H-吡唑-1-基)乙-1-酮(2f).Yield 56%, m.p.279-281 ℃.1H NMR(DMSO-d6,300 MHz)δ:2.30(s,3H,COCH3),2.34(s,3H,Ph-CH3),3.15(dd,1H,J1=4.7 Hz,J2=18.3 Hz, CH2-Ha), 3.86(dd, 1H,J1=11.5 Hz,J2=18.3 Hz,CH2-Hb),5.60(dd,1H,J1=4.7 Hz,J2=11.5 Hz,CH),7.06-7.70(m,10H,Ar-H),9.70(s,1H,OH).MSm/z345(M+1)。
1-(5-(6-羟基萘-2-基)-3-(4-甲氧基苯基)-4,5-二氢-1H-吡唑-1-基)乙烷-1-酮(2g).Yield 55 %, m.p.230-232 ℃.1H NMR (DMSO-d6, 300 MHz)δ: 2.30 (s, 3H, COCH3), 3.17 (dd, 1H,J1= 4.1 Hz,J2= 18.2 Hz, CH2-Ha), 3.79 (s, 3H, OCH3), 3.85(dd, 1H,J1= 11.4 Hz,J2= 18.2 Hz, CH2-Hb), 5.60(dd,1H,J1=4.1 Hz,J2=11.4 Hz,CH),6.99-7.75(m,10H,Ar-H),9.67(s,1H,OH).MSm/z361(M+1)。
1.4 PTP1B 体外抑制试验
合成化合物对PTP1B 的抑制活性使用对硝基苯磷酸酯(pNPP)作为底物来测定。pNPP 被PTP1B 水解后会产生去磷酸的对硝基苯酚,其在405 nm 处显示明显的吸收峰。经过PTP1B 处理后的p-硝基苯酚含量表示PTP1B 酶的活性,进而表示化合物对PTP1B 酶活性的抑制作用[24]。具体操作方法如下。将2 mmol/L 的pNPP 和PTP1B(0.05 ~0.1 μg)加入含有50 mmol/L 柠檬酸(pH为6.0)、0.1 mol/L 氯化钠、1 mmol/L 乙二胺四乙酸(EDTA)和1 mmol/L 二硫苏糖醇(DTT)的缓冲液,缓冲液中不加或加入不同浓度的受试化合物。在37℃孵育30 min 后,用10 mol/L NaOH终止反应。测定在405 nm 处的吸收度来获得化合物对酶活性的抑制情况。使用Prism 软件对化合物5 个以上浓度下的酶抑制情况进行拟合,并计算ⅠC50值。
1.5 分子对接
为了探究羟基萘查尔酮与PTP1B 的分子对接情况,验证其是否按照实验的设计思路与PTP1B活性位点产生了更好的结合,选择了代表性化合物1d,将其与PTP1B 分子进行虚拟对接。PTP1B酶的3D 结构下载自蛋白数据库(Protein Data Bank,ⅠD: 1NL9)。采用Discovery Studio 2019 版 对蛋白分子进行除水加氢处理,活性位点选择原配体结合位点。化合物1d 结构采用Chem3D Ultra 16.0 软件绘制后,采用Discovery Studio 分子处理模块进行能量最小化,然后将处理后的化合物结构与蛋白进行分子对接。分子对接结束后,按照结合能和docking score 进行排序,取得分最高的结合模式进行分析[25]。
2 结果与讨论
2.1 化学合成
化合物1a-1g 和2a-2g 的合成路线如图3 所示。通过苯乙酮(或者取代苯乙酮)与对6-羟基-2-萘甲醛在NaOH 碱性条件下的克莱森-施密特缩合反应合成了羟基萘查尔酮(1a-1g)。将羟基萘查尔酮与乙酰肼在乙醇中回流,制得化合物2a-2g[26]。通过核磁共振氢谱和质谱确证了化合物1a-1g 和2a-2g 的结构。在核磁共振氢谱中,化合物1a-1g 在化学位移7-8 范围出现的两个耦合常数为15.5 ~16.0 的双重峰,直接说明了α,β-不饱和酮的形成,并且因为耦合常数大于12,说明生成的萘查尔酮为E 构型。化合物2a-2g 的氢谱中,出现了三簇dd 峰,并且其耦合常数一一对应,说明了加氢吡唑环的形成。结合质谱信息,可以确证合成的化合物为目标结构。
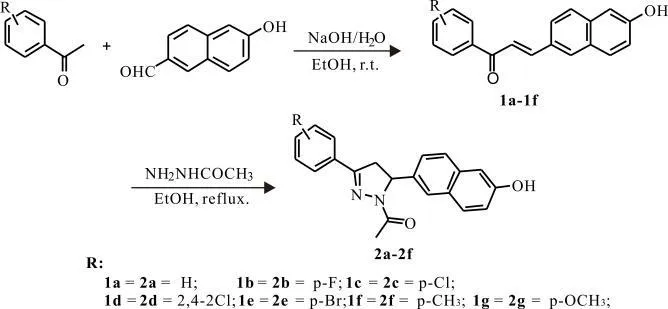
图3 目标化合物1a-1g 和2a-2g 的合成路线Fig.3 The synthesis route of the target compounds(1a-1g and 2a-2g)
2.2 化合物的PTP1B 抑制活性
首先在浓度为20 μg/mL 的情况下,对合成化合物的PTP1B 抑制活性进行了初步的测定。如表1所示,所有化合物都表现出不同程度的PTP1B 抑制活性,抑制率在31.16%~96.74%范围。其中,化合物1d、1g、2c 和2f 表现出明显的抑制活性,抑制率高于90%。通过比较这些化合物的抑制活性,羟基萘查尔酮在环合前后的活性似乎没有明显规律。在羟基萘查尔酮系列中(1a-1g),2,4-二氯取代化合物(1d)和4-甲氧基取代化合物(1g)活性最好。而在羟基萘查尔酮环合系列中(2a-2g),4-氯取代化合物(2c)和4-甲基取代化合物(2f)的PTP1B 抑制活性更强。
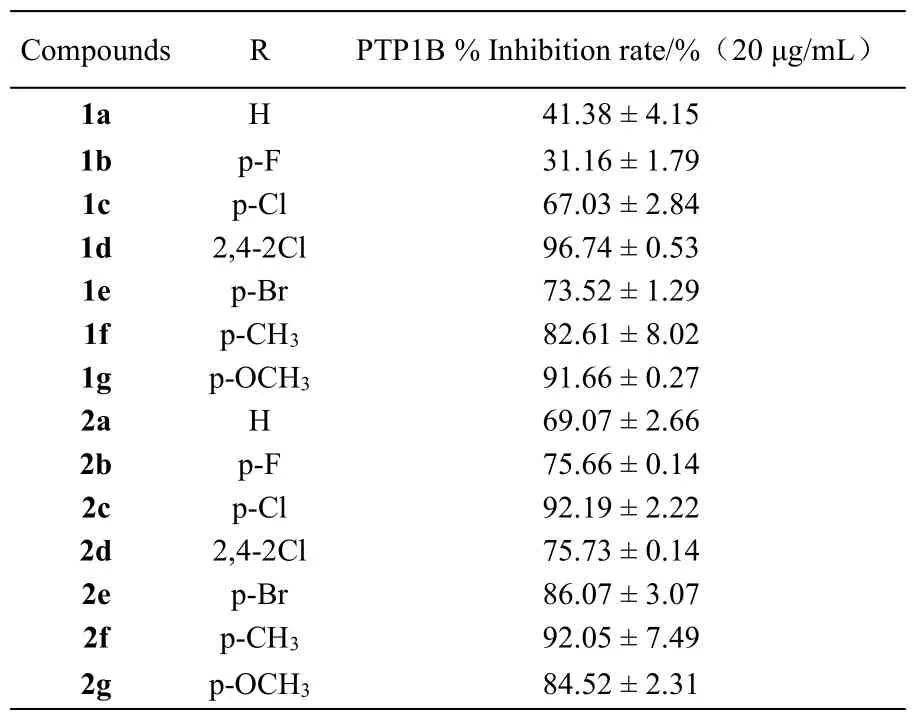
表1 1a-1g and 2a-2g 在20 μg/mL 浓度下的PTP1B 抑制活性Table 1 The preliminary inhibitory activity of compounds 1a-1g and 2a-2g against PTP1B
将化合物1a 和1d 在不同浓度下进行抑制率测定,得到了它们的半数抑制浓度ⅠC50。在相同条件下测定了PTP1B 抑制剂熊果酸的ⅠC50,用作阳性对照[27]。如表2 所示,化合物1d、1g、2c 和2f的ⅠC50值分别为1.91、8.59、7.38、4.64 μM。这个系列中最有效的化合物,化合物1d 的PTP1B 抑制活性高于阳性对照熊果酸的抑制活性,也要明显高于本实验的先导化合物(Ⅰ和ⅠⅠ)。

表2 化合物1a、1g、2c 和2f 的定量PTP1B 抑制活性Table 2 The quantifed inhibitory activity of the selected compounds 1a、1g、2cand 2f against PTP1B
2.3 化合物1d 与PTP1B 蛋白的分子对接
为了验证本实验设计的合理性,探究目标化合物是否比先导化合物(Ⅰ)更好地与PTP1B 活性位点结合,将化合物1d与PTP1B 酶(蛋白质数据库,PDBⅠD:1NL9)进行了分子虚拟对接。化合物1d与PTP1B 蛋白的氨基酸互作用如图4 所示。化合物1d与PTP1B 蛋白酶中的TYR46、VAL49、PRO180、PHE182、GLY183、ARG221 氨基酸残基产生结合。其中,化合物1d的羟基与PTP1B 氨基酸残基PRO180 形成氢键而产生重要结合力。此外化合物结构中的萘环与氨基酸残基PHE182 和ARG221 等形成疏水性和Pi 健相互作用,苯环上的两个氯原子也分别与TYR46 和VAL49 产生疏水性结合,进一步提高了化合物与受体的结合力。化合物1d和先导物I的分子对接docking score 分别为63.89 和57.38,说明化合物1d比先导物I能够更好的与PTP1B 结合,这可能是其产生更高PTP1B 抑制活性的主要原因。因此,本实验的萘环设计,从提高受体结合力的角度提高了PTP1B的抑制活性,这为后续更高PTP1B 抑制活性的衍生物发现提供了重要方向。
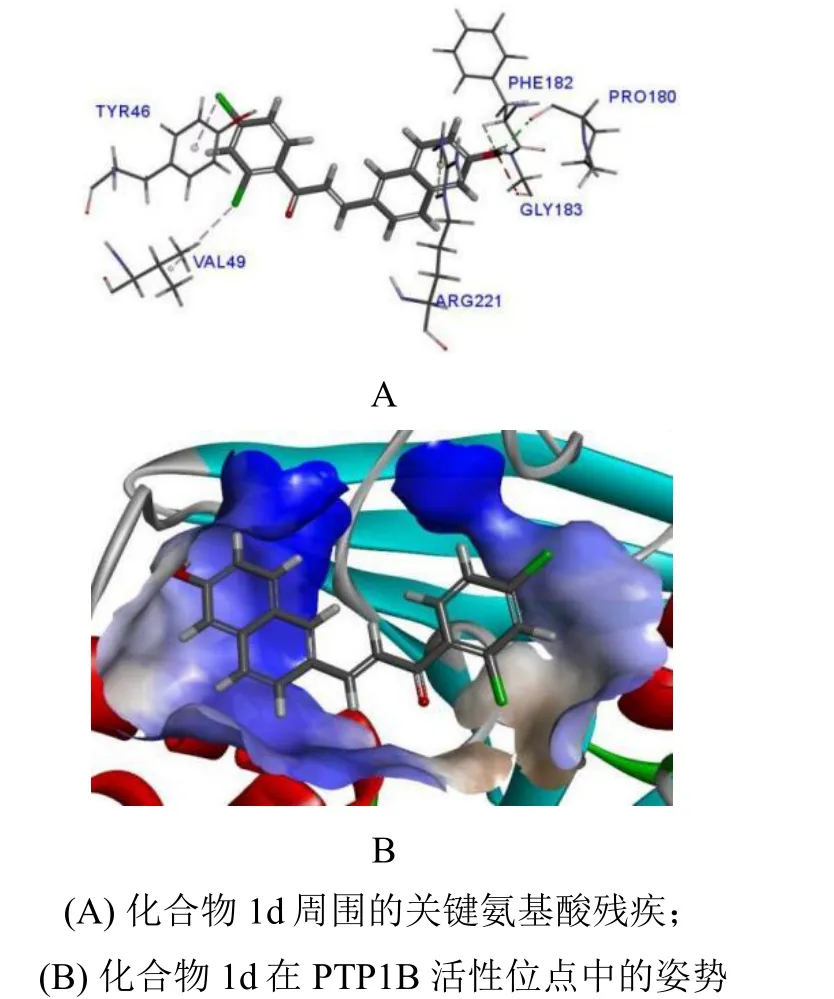
图4 化合物1d 与PTP1B 的结合模式Fig.4 The binding modes of compound 1d in the PTP1B catalytic pocket
3 小结
总之,本实验合成了一系列羟基萘查尔酮及其环化衍生物,并评估了它们对PTP1B 酶的抑制活性。实验中获得了4 个ⅠC50在微摩尔级别的PTP1B 抑制剂。分子对接分析证实了羟基萘查尔酮与PTP1B 的相互作用,并提示萘环等更大的亲脂性基团有助与提高该骨架的PTP1B 抑制活性。这些结果为开发具有良好细胞渗透性和生物利用度的新型PTP1B 抑制剂提供了新的方向。
