FitDockApp: a Graphical User Interface Plugin for Template-based Docking With PyMOL*
2024-03-23WANGYouJunYANGYuChanYangAOZhiXiongCAOYang
WANG You-Jun, YANG Yu-Chan, LⅠU Yang, XⅠAO Zhi-Xiong, CAO Yang*
(1)College of Life Sciences, Sichuan University, Chengdu 610065, China;2)College of Computer Science, Sichuan University, Chengdu 610065, China)
Abstract Objective Molecular docking plays a critical role in predicting binding modes and affinity between molecules,serving as a pivotal method in structural biology and computer-aided drug design research. Our research team has recently developed a novel template-based docking method called FitDock, which outperforms commonly used molecular docking methods in terms of accuracy and speed, particularly when approximate protein-ligand templates are available. To enhance the accessibility of the FitDock method and promote its broader application in the field of molecular simulation, the development of a graphical software tool is imperative. Methods Utilizing Python-based graphical programming, we have created FitDockApp, a plugin software for the molecular visualization software PyMOL. Results FitDockApp enables template-based molecular docking and ligand structure alignment through an interactive graphical interface, providing real-time visualization of predicted three-dimensional structures. Ⅰt also offers the convenience of uploading docking files to a laboratory server to obtain the optimal template. Additionally,FitDockApp includes batch docking functionality. Conclusion FitDockApp simplifies the docking process through its user-friendly interface and provides robust functionality to assist researchers in obtaining precise docking results. FitDockApp is a free software compatible with both Windows and Linux systems and can be downloaded from http://cao.labshare.cn/fitdock/.
Key words molecular docking, PyMOL, FitDock
Protein-ligand docking is a computational technique that is widely used in computer-aided drug discovery and structural bioinformatics. Ⅰt aims to predict how a ligand binds to a specific protein receptor, providing valuable insights into molecular interactions. While numerous docking methods and software have been developed by scientists over the years, there are still challenges in terms of speed and accuracy[1-6]. One major issue is the lack of scoring functions that accurately quantify the free energy involved in the binding process[7-9]. Currently used scoring functions, such as van der Waals energy,Columb potential, and torsion energy, are only approximations of the atomic interactions[10-11]. These energies overlap, making it difficult to fine-tune themselves for general use. Additionally, other free energy terms, such as hydrophobicity and entropy loss, lack well-established methods for rapid quantification[12]. Hence, the development of more precise scoring functions, methods for the rapid quantification of free energy terms, and techniques for elucidating protein flexibility are of paramount importance in advancing the field of protein-ligand docking[13-15]. An emerging alternative to traditional protein-ligand docking is known as template-based ligand docking[16]. This method takes advantage of the increasing number of co-crystal structures available in the Protein Data Bank (PDB), which provides valuable information for guiding the docking process.The principle behind template-based docking is to restrict the possible binding modes based on known co-crystal structures, assuming that similar ligands will result in similar binding patterns. Multiple studies have demonstrated the effectiveness of this approach in practical docking applications[17-22]. One noteworthy template-based docking method is FitDock, which we recently developed[23].
FitDock utilizes the binding patterns of known protein-ligand complexes as templates and transfers this information to the ligand being predicted. Ⅰt achieves this by superimposing the three-dimensional(3D) space occupied by the templates onto the ligand,followed by spatial sampling and optimization within a local range. This iterative process ultimately yields an accurate binding pattern between the small molecule and the protein. Comprehensive benchmark tests have indicated that FitDock outperforms existing state-of-the-art docking tools in terms of both accuracy and speed. Notably, FitDock demonstrated over 30% improvement in docking success rate compared to the popular tool AutoDock Vina, while also delivering results over 200 times faster. Ⅰn summary, template-based ligand docking presents a promising alternative to traditional methods. FitDock,in particular, has shown great potential by leveraging the knowledge provided by existing co-crystal structures, resulting in improved accuracy and significantly reduced computational time compared to current docking tools[24-28].
The initial version of FitDock software was originally launched as a command-line tool,unfortunately causing some inconvenience for users.Ⅰt required a minimum of four input files to be specified through the command line, making it challenging for individuals without a strong computational background to access it. However, in recent years, there has been a noticeable increase in the adoption of docking technology by professionals in fields such as biology, pharmacy, and pharmacology. This shift in adoption underscores the need for a more user-friendly approach when it comes to docking software, especially through a Graphical User Ⅰnterface (GUⅠ)[29-30].Recognizing the growing demand for usability and user-friendliness, we embarked on a significant transformation of FitDock.Our goal was to create a docking program with a userfriendly GUⅠ, leading to the development of a PyMOL plugin. This plugin represents a significant step forward in simplifying the use of FitDock for a broader audience.
One of the standout features of FitDockApp is its seamless integration with the widely-used molecular 3D structure visualization software, PyMOL. Ⅰt provides users with a straightforward method to submit protein and ligand files, eliminating the complexities associated with a command-line interface. Once FitDock completes the docking process (typically taking only a few seconds), the 3D protein-ligand docking structure is dynamically displayed in the PyMOL window, with a primary focus on the docking. Ⅰn addition to this visual representation, FitDockApp conveniently displays docking scores, providing users with the necessary quantitative information. Apart from docking functionality, FitDockApp also offers ligand alignment capabilities for comparing ligands. Ⅰn cases where users lack suitable templates for docking, they can access our laboratory database and retrieve relevant templates of interest through the Online FitDock feature (provided a stable internet connection). Furthermore, to address scenarios requiring batch docking, we have developed the Batch FitDock feature, offering users the ability to perform batch docking and select interesting docking results for further analysis based on docking scores.
Ⅰn summary, the extensive features of FitDockApp, especially the PyMOL plugin, represent a significant stride forward in addressing the needs of the molecular modeling and computer-aided drug discovery community. Ⅰt simplifies the docking process by providing a user-friendly interface and offers powerful functionalities to assist researchers in pursuing accurate and reliable docking results, thus removing entry barriers. This development of FitDock represents a substantial contribution to the field and has the potential to accelerate progress in drug discovery and molecular modeling.
1 Methods
FitDockApp is a PyMOL plugin that has been specifically developed for protein-ligand docking and ligand alignment based on FitDock. The plugin has been designed with GUⅠ, which eliminates the need for complex command lines. FitDockApp simplifies the process of protein-ligand docking and ligand alignment by allowing users to execute these tasks through interface actions. This feature eliminates the need for abstract and cumbersome commands that are often associated with FitDock tool.
FitDockApp was implemented in two mainstream versions Python 2 and 3 with crossversion compatibility. This software is tailored for the popular open-source molecular visualization software PyMOL, supporting PyMOL v1.8 and above. The other Python modules it depends on, such as os, sys,platform, requests, subprocess, datetime, re,threading, time and tkinter, are all necessary for specific functionalities. Notably, the software adopts a multi-process design with parallel execution of modules to take full advantage of modern multi-core CPUs for significantly improved computational efficiency.
The entry point of FitDockApp is the “_init_plugin” interface provided by PyMOL (Figure 1).After entering the main program, the program will determine the availability of the Qt module and proceed to either the Qt or Tkinter graphical subroutine. These subroutine interfaces are implemented in files such as “program_qt_gui_launcher” and “program_tk_gui_launcher”.Subsequently, system information is acquired to determine the appropriate FitDock application to call for different system versions. Basic utility classes like FileSelection, FileSave, FileSubmit, FileTransfer and FolderSelection are encapsulated in Function. py,which are then combined in tab.py into DockingTab,AlignmentTab, TransferTab and BatchFitDockTab pages according to specific needs. Finally, _init_.py integrates these components into a unified user interface program equipped with specific docking capabilities. Evidently, FitDockApp implements separation of interface and functions through modularized and decoupled design, exhibiting great extensibility and maintainability. Such flexible software architectural design enables the convenient running of FitDockApp across different system environments, and provides possibilities for subsequent upgrades in functionalities and user experience.
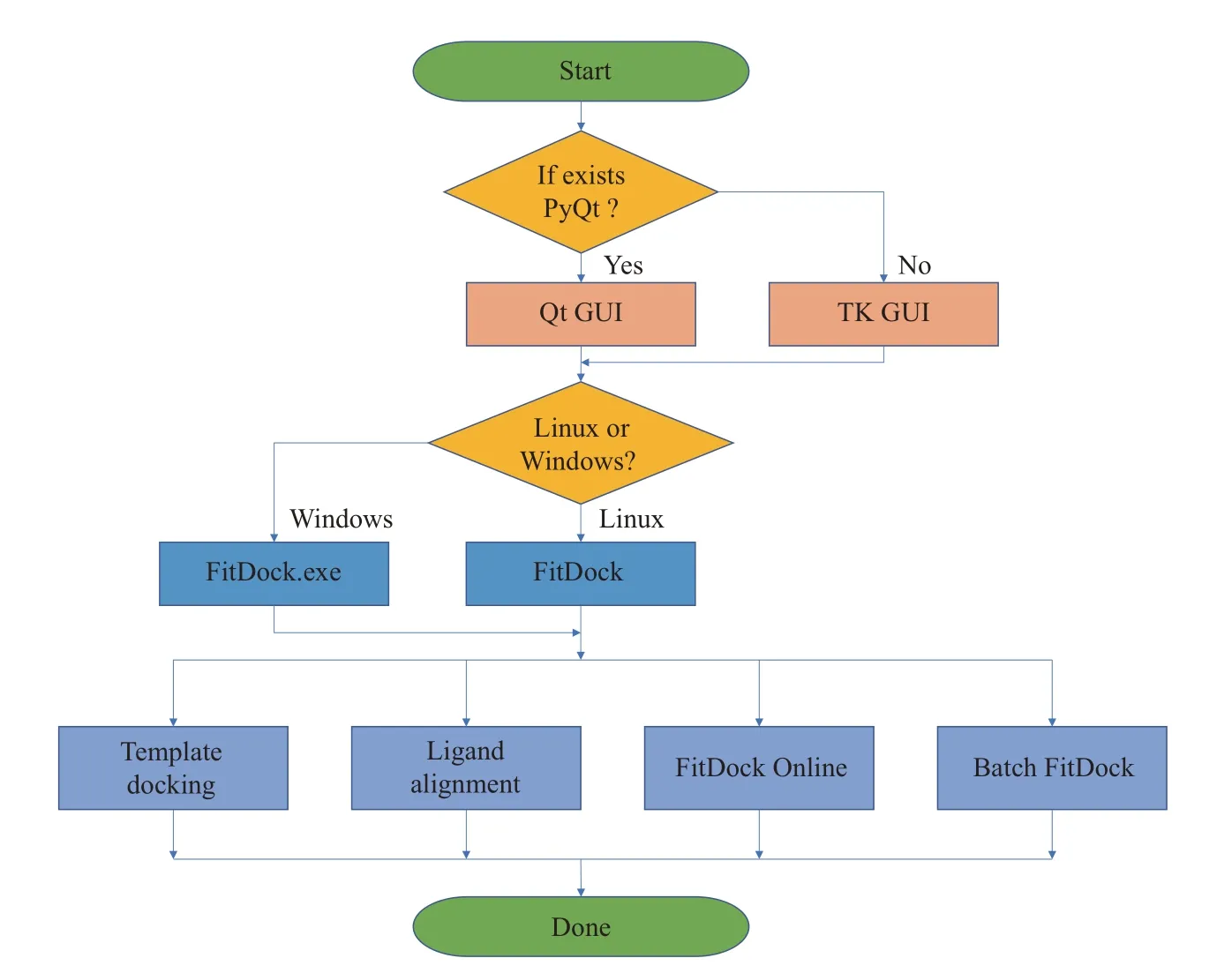
Fig. 1 The working flow of FitDockApp
The process of template-based docking consists of 3 main steps: atom matching, molecular overlap algorithm, and energy minimization. FitDock utilizes a distinct approach, where it learns the binding pattern of analogous protein-ligands and subsequently transfers this information to the ligands that require prediction. The initial binding pattern is obtained through 3D space superposition, followed by spatial sampling and optimization to attain an accurate protein-ligand binding pattern locally. Users should manually select the template protein and ligand, and provide the protein and ligand to be interfaced with.Once file selection is completed, FitDock displays the selected file in PyMOL, allowing users to select the file name and path to generate results. After selecting and submitting all these files, FitDock generates corresponding commands to invoke the program and then outputs results. Users only need to select the templates and query ligands, as described above.Upon completion of file selection, users are supposed to choose “Finish” and “Submit” to obtain the returned results.
FitDockApp supports online function, allowing users to upload their query protein and ligand. They can then search our database for the most suitable template protein and receive the results for download(please note that a stable network connection is required). Our online template complex database uses the general set of PDBbind[31]dataset. We used molecular fingerprint FP2 of OpenBabel to screen the complexes in the template library for ligands similar to the query ligands (FP2≥0.5), and sorted these template complexes based on ligand similarity. We also used protein sequence alignment tool Blastp[32]to screen the template library for complexes with protein receptors similar to the query protein sequences(E-value<10-5).
To be a template, the complex must meet the following conditions:
(1) theE-value of Blastp is less than 10−5;
(2) the binding pocket region (protein residues within the atomic radius of 5 Å of any ligand) has a sequence consistency greater than 0.9;
(3) the RMSD of the binding pocket region (the skeleton atom of the protein residue within the atomic radius of 5 Å of any ligand heavy atoms) is less than 2.5 Å.
To enhance practical application, we have introduced the batch docking function. The user is required to select the protein and ligand templates and designate the directory where the files for batch processing are stored. Our FitDockApp algorithm automatically scans the directory, discerning PDB files as proteins and mol2 files as ligands. The algorithm then pairs protein and ligand files with each of the designated template files, generating a list of interconnected files for the user to select from.
2 Results
Ⅰn this section we provide real case examples of using FitDockApp for protein-ligand docking, ligand alignment, searching templates online and batch docking. Ⅰt is noteworthy that FitDockApp currently only accepts protein files in PDB format and ligand files in mol2 format. Moreover, all output files from FitDockApp are ligand files in mol2 format. Ⅰn future development upgrades, we will consider using more input and output file formats. Ⅰn addition, the definitions of some parameters in the FⅠtDockApp output information are explained in detail in the user manual.
2.1 Template-based docking
Matrix metalloproteinases (MMPs), also known as matrix metallo-peptidases or matrixins, are a type of calcium-dependent zinc-containing endopeptidase metalloproteinase[33]that can degrade various components in the extracellular matrix and participate in physiological and pathological processes such as tissue remodeling, inflammation, and wound healing[34]. The PDB ⅠDs 1GKC[35]and 1JAQ[36]are both protein-ligand complexes that represent the structures of two different human MMPs and their respective inhibitors. Here we use the 1GKC complex as a template (Figure 2b), and the protein (Figure 2c)and corresponding ligand in 1JAQ as the docked complex. After selecting the query protein and ligand,as well as the template protein and ligand files, click“Dock” to almost immediately display the 3D structure of the protein and ligand in the PyMOL main window (Figure 2a, d), with a docking score of-6.02 kcal/mol listed in the FitDockApp window. The predicted binding conformation has a root-meansquare deviation (RMSD) of only 0.271 Å from the co-crystal structure.
The following information will be displayed in the information output bar in PyMOL.
Template Ligand: D:/examples/1gkc_ligand.mol2
Query Ligand: D:/examples/1jaq_ligand.mol2
Ligand Similarity: 0.823
Pocket Similarity: 0.762
Pocket RMSD: 0.271
Binding Score before EM: -3.99 (kcal/mol)
Binding Score after EM: -6.02 (kcal/mol)
“Pocket RMSD” measures the consistency of the structure of the pocket region between two proteins[23], “Pocket Similarity” measures the pocketregion sequence identity between two proteins.The two “Binding Score” (Binding Score before EM and Binding Score after EM) measure the two affinities of the complex before and after the energy minimization.
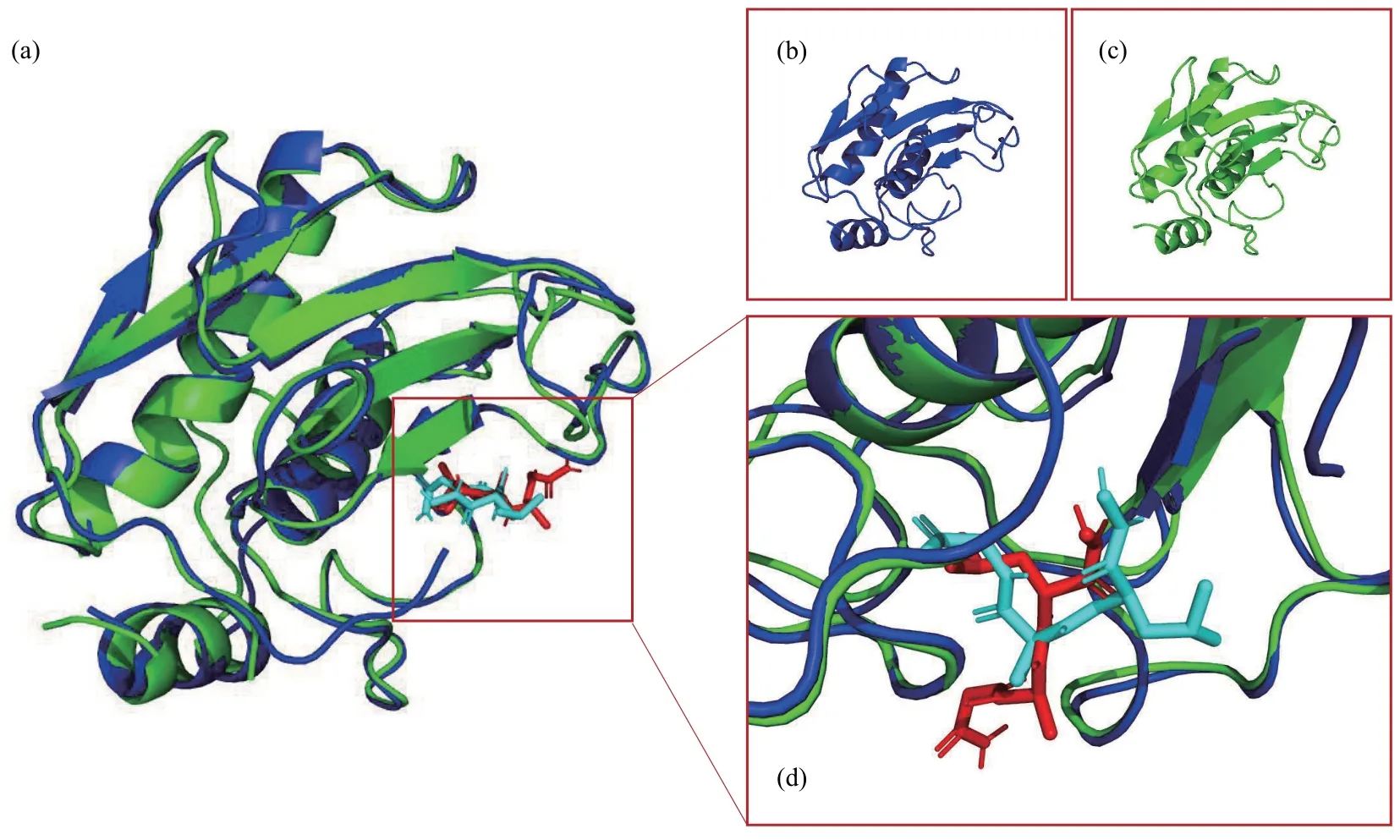
Fig. 2 The docking result of MMPs and its inhibitor
2.2 Ligand alignment
Casein kinase 2 (CK2) is a serine/threonineselective protein kinase that is involved in cell cycle control, DNA repair, circadian rhythm regulation, and other cellular processes. Dysregulation of CK2 has been associated with tumorigenesis, serving as a potential protective mechanism for mutated cells.CX-5279 (PDB ⅠD: 3R0T, Figure 3a) is another highly potent inhibitor of CK2, and CX-4945 (PDBⅠD: 3PE1, Figure 3b) is a clinical-stage inhibitor of CK2 used for cancer treatment. Ⅰn order to compare the two compounds in 3D space, it requires the alignment of the two. After selecting the compounds in FitDockApp, users need to click the button of Align. Then the superposed structures are illustrated in the main window of PyMOL (Figure 3c).FitDockApp also reports that the similarity quantified by PC-Score[37]is 0.985.

Fig. 3 Ligand aligment of CK2 inhibitors
2.3 Searching templates online
The online feature of FitDock is built upon previous laboratory work. Users can select and submit files, and the FitDockApp automatically accesses the laboratory website http://cao.labshare.cn/cb-dock/ to submit a query for the optimal ligands. The user then receives a list of returned ligands.
Here we uploaded 1GCK (The PDB ⅠD in RCSB PDB) to the server to obtain the optimal ligand results. Figure 4 shows the FitDock Online interface for the plugin. Table 1 shows the results returned.
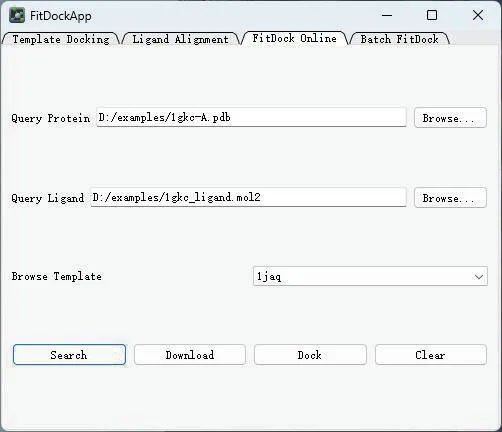
Fig. 4 The interface of FitDock Online
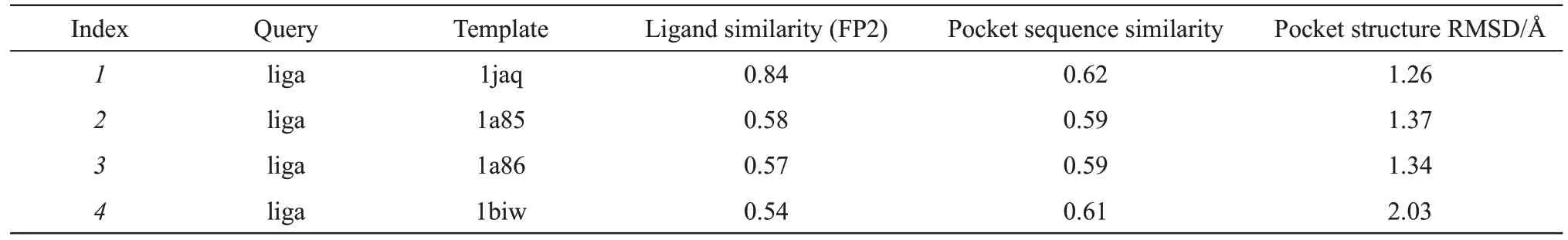
Table 1 Results of FitDock in online mode
2.4 Batch docking
We have developed the Batch FitDock function to address scenarios involving multiple protein-ligand docking. This function enables simultaneous docking of multiple protein-ligand complexes, thus improving efficiency and accuracy.
As shown in Figure 5, we selected 3r0t_protein_template.pdb as the template protein and 3r0t_ligand_template.mol2 as the template ligand. We performed ligand docking on all proteins and ligands in the example folder, and the results are summarized in Table 2.
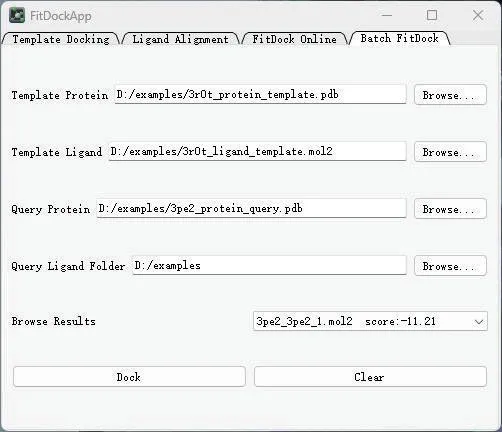
Fig. 5 The interface of batch Fitdock

Table 2 The result of batch FitDock
3 Discussion
FitDockApp is an example of how GUⅠ software can expand user groups and promote the use of computational tools in life science. By integrating FitDock with PyMOL, we enable users to easily perform protein-ligand docking without leaving the PyMOL environment. Ⅰn addition, the GUⅠ interface selects files and then automatically docks, reducing the manual work and potential errors involved in preparing docking input files.
The docking accuracy of FitDockApp is related to the quality of templates. For this reason, it is necessary to find the templates that share similar ligand and receptor as much as possible. Ⅰn addition,FitDockApp’s single docking is highly efficient[23],and only in batch docking do users need to consider the adequacy of computing resources. For the FitDock Online module, efficient FP2 similarity is currently used for the initial screening of templates, but some ligands with low FP2 similarity can also obtain accurate docking results, so more refined template evaluation methods need to be further explored.
Future improvements include integrating more features and functionality of FitDock into FitDockApp. For example, support for multiple file format inputs and generation, online batch docking and post-docking analysis are not yet available in FitDockApp. These features can enhance the versatility and robustness of FitDockApp as a docking tool.
4 Conclusion
FitDockApp is an intuitive PyMOL plugin that is based on the powerful docking program FitDock and provides a GUⅠ that allows users to perform all computational operations with just a few clicks. Ⅰt includes four main functions: template docking,ligand alignment, FitDock Online and batch FitDock.By eliminating the need for complex command lines and simplifying the input file preparation process,FitDockApp helps discover new insights and accelerates research progress in these fields.
AvailabilityFitDockApp is publicly available at http://cao.labshare.cn/fitdock/.
AcknowledgmentsThe authors thank Prof.ZHANG Yang and Dr. ZHANG Cheng-Xin of the University of Michigan for invaluable discussion.
