DNA聚合酶θ:易错的多功能DNA末端修复分子*
2024-03-23陈国江冯健男石艳春郑源强
王 瑶 陈国江 冯健男 石艳春** 王 晶** 郑源强**
(1)内蒙古医科大学,内蒙古自治区分子生物学重点实验室,呼和浩特 010058;2)军事科学院军事医学研究院毒物药物研究所,抗毒药物与毒理学国家重点实验室,北京 100850)
Polθ(DNA polymerase theta),又被称为DNA聚合酶θ,是DNA 聚合酶 A 家族成员之一,参与DNA 双链断裂(DNA double-strand breaks,DSB)修复过程[1-4]。Polθ 具有独特的结构,是目前已知真核生物中发现的唯一含有解旋酶活性的聚合酶,同时也是一种易出错的聚合酶。机体内DSB 发生后常见有4种修复途径(图1),即经典非同源末端连接 (classical-non homologous end joining,C-NHEJ)、同源重组(homologous recombination,HR)、单链退火(single-strand annealing,SSA)和选择性末端连接(alternative-end joining,Alt-EJ)。当主要的DNA 修复通路缺陷时(如HR 通路),细胞会依赖Polθ 介导的Alt-EJ 通路来修复损伤的DNA,以维持基因组稳定性[5]。然而,由于Polθ 具有低保真性的特点,Alt-EJ 修复常常会出错。此外,癌症中常可见过度表达的Polθ,抑制Polθ 与各种DNA 修复基因的联合缺失会导致合成致死(synthetic lethality,SL),包括已知的癌症驱动因子(如BRCA1/2)[6]。近年来,小分子靶向化疗药成为抑制肿瘤生长的热点,但是脱靶效应和耐药性成为困扰其临床应用的主要问题,尤其是对聚ADP 核糖聚合酶抑制剂(poly-ADP-ribose polymerase inhibitor,PARPi)的耐药性。研究发现,在HR 缺陷肿瘤细胞中阻断Polθ 活性可逆转PARPi耐药,这使Polθ在癌症治疗中成为一个非常有前途的治疗靶点[7]。目前,已经报道了多种Polθ抑制剂,如新生霉素(Novobiocin,NVB)[7]、ART558[8]和RP-6685[9]等。这些抑制剂的研制为治愈BRCA1/2突变的肿瘤提供了新的方向。此外,通过减少Polθ介导的Alt-EJ修复途径还有助于提高HR 修复效率,增加外源基因靶向整合(target integration,TⅠ)几率,使Polθ 抑制剂又具有新的潜在应用价值。本文综述近几年关于Polθ 分子功能及其参与Alt-EJ通路修饰机制的研究进展,为深入了解该领域提供思路。
1 Polθ的结构特征及功能
Polθ 由POLQ基因编码,由2 590 个氨基酸组成,分子质量为290 ku,主要参与Alt-EJ 途径对DSB进行修复。POLQ基因在包括植物和原生生物在内的多细胞真核生物中广泛存在。然而,在包括酵母在内的真菌中,并没有发现POLQ类似基因[10]。Polθ 结构独特,是哺乳动物细胞中唯一包含解旋酶功能的DNA 聚合酶。该分子主要包含3个结构域(图2):氮(N)端含保守序列的解旋酶样结构域(helicase-like domain)、碳(C) 端的DNA 聚合酶结构域(polymerase domain)以及连接这两个区域的一段无序序列的中心区域(central domain)[10]。
N端是超家族2解旋酶样结构域(superfamily 2 helicase domain,Polθ-Hel),具有DNA依赖的ATP酶活性,其在复制进化过程中基本保持不变。Polθ-Hel通过取代复制蛋白A(reputation protein A,RPA)来阻止HR 修复。Polθ-Hel 可以在长单链DNA (single-stranded DNA, ssDNA) 上促进Alt-EJ[11]。
C 端是DNA 聚合酶A 家族结构域(family-A polymerase domain,Polθ-Pol),包括3 个插入氨基酸环,促使短单链DNA 引物的相互作用、退火和延伸[12-13]。在Alt-EJ 中,Polθ-Pol 可以作为末端转移酶或催化经过热处理的序列的模板延伸。除了双链断裂修复外,该聚合酶还可以作为5'-dRP裂解酶在碱基切除修复(base excision repair,BER)中发挥作用,并对紫外线损伤进行跨损伤合成(translesion synthesis,TLS)。Alt-EJ 在连接退火的DSB末端之前,利用Polθ-Pol填充DNA间隙。
中心区域由一个外显子跨越了整个非结构化的中心氨基酸序列[14]组成,虽然在大小和跨物种序列方面保守性较差,但可将Polθ-Hel 和Polθ-Pol 连接在一起,其中包含与RAD51 结合模块。另外,该中心区域对底物选择至关重要,它能自动抑制短ssDNA上的Polθ的活性。

Fig. 2 The structure of Polθ图2 Polθ结构示意图
2 Polθ与DSB修复通路
DSB 是由拓扑异构酶抑制剂、电离辐射、紫外线照射、停滞的DNA 复制分叉以及CRⅠSPR/Cas9 等诱导,可造成DNA 复制或转录停滞,进而引发基因重排、细胞坏死甚至细胞减数分裂过程中断和促进癌症发生发展等。现阶段发现,DSB 断裂之后有4 种主要修复途径(图1),即C-NHEJ、HR、SSA和Alt-EJ。大多数DSB是由C-NHEJ介导修复的,可以发生在整个细胞周期中,通过在断裂位点引入小DNA 片段的插入和删除以及染色体易位来直接连接DNA 末端[15]。在S 和G2 期,HR 使用姐妹染色单体或同源染色体作为模板进行DNA合成,是目前已知唯一精确的DSB 修复途径[16]。SSA 是DSB 修复的第三种途径,消除了重复序列之间的DNA 片段[17],通常导致大量的缺失和DNA重排,对基因组完整性有害。当主要的DNA修复通路缺失时,细胞会依赖替代通路,即Alt-EJ, 也称微同源性介导的末端连接(microhomology-mediated end-joining,MMEJ) 和聚合酶介导的末端连接(theta-mediated end joining,TMEJ)[5]。这种修复途径具有内在的诱变性,断裂位点通常因插入和删除而受损。DSB 修复通路中,4 种通路都至关重要,除HR 介导靶向整合外,其他通路都介导随机修复。
2.1 Polθ介导选择性末端连接(Alt-EJ) 修复通路
Alt-EJ 包括许多由5'-3'的切除因子,如PARP1[18]、 Polθ[19]、 CtBP 相互作用蛋白(carboxy-terminal binding protein,CtⅠP)[20],MRN复合体(MRE11-RAD50-NBS1,MRN)[21],DNA连接酶1(DNA ligase Ⅰ,LⅠG1)和DNA 连接酶3(DNA ligase Ⅲ,LⅠG3)[22]等。Polθ 在Alt-EJ 通路中起着核心作用[23],可以延长3'单链和双链DNA的―OH末端,跨越脱嘌呤嘧啶位点或以不依赖模板的方式错配。Alt-EJ 通路中(图1),首先由PARP1募集到DSB处并激活MRN/CtⅠP复合物[24],以暴露断裂位点的微同源序列[25-28]。MRN/CtⅠP 处理DNA末端产生3' DNA悬垂,Polθ结合由DSB的5'-3'重剪切生成的长单链DNA(ssDNA)悬臂,并通过与具有2~6个碱基对的微同源序列退火,将它们用作DNA 合成的引物[29-30],促使修复过程初始阶段形成的中间DNA 结构稳定。经过内切酶等其他酶对过渡结构的处理后,最后由LⅠG3或LⅠG1来促进最终DNA间隙的融合[31]。其中,PARP1依赖的Polθ 向DSB 位点的募集是这一途径的关键步骤[6,19]。
2.2 Polθ参与DSB修复以外的修复途径
Polθ 除了在DSB 修复途径中发挥作用,还参与 BER、 DNA 链间交联 (DNA interstrand crosslinks,ⅠCLs)修复、TLS等过程,并且可能是特定类型DNA 损伤的唯一可用途径[28]。由于Polθ-Pol有较弱的5'-脱氧核糖磷酸裂解酶活性,因此,通常认为Polθ在BER中起作用[32]。DNA单链断裂在遇到复制分叉时转化为DSB 也可能依赖于Polθ 的修复[33]。在果蝇和线虫中,Polθ 参与的ⅠCLs 和Polθ-Hel 活性对果蝇对氮芥抗性至关重要[34-35]。另外,Polθ 对G4 四重体结构的修复不可或缺,防止以小缺失为代价的基因组重排[5]。Bela等[36]证明,Polθ-Pol 可以催化一种独特的诱变间隙填充反应,称为微同源介导的DNA 间隙跳跃(microhomology-mediated gap skipping,MMGS),该反应通过微同源性退火导致间隙填充反应中缺失的形成,可能是产生Polθ 依赖性基因组瘢痕的潜在机制。MMGS可以绕过多种病变,不需要与病变相对的核苷酸掺入,在HR缺陷细胞中引起Polθ活性的突变特征。Yi 等[37]证明,Polθ 的TLS 活性在高线能传递辐射诱导的复杂DSB中起着关键作用。
3 Polθ与基因组稳定性
DNA 损伤反应 (DNA-damage response,DDR)通过内源性和外源性DNA 损伤并激活特定的DNA 修复途径来保持基因组的完整性。正常组织细胞中,ⅠCL和DSB修复缺陷可以导致染色体不稳定,并抑制DNA 复制和转录,进一步导致相关疾病的发生。Ceccaldi 等[6]利用小干扰RNA 敲低Polθ,发现Polθ 减少与DSB 形成增加、复制叉的不稳定以及对某些基因毒性因子的敏感性增强有关,因此,推测Polθ 在基因组稳定性中起着至关重要的作用。另有研究发现,POLQ基因缺失的正常组织细胞甚至肿瘤细胞对外界因素,包括过氧化氢、γ射线等变得敏感[38-39],进而引发基因不稳定等突变事件发生。Polθ除了在哺乳动物基因组稳定中起作用,在植物中同样起着重要作用[40]。Plecenikova 等[41]发现,Polθ 缺失增加苔藓和单细胞衣藻对博莱霉素(用于治疗癌症的药物,会引起DNA 双链断裂)的敏感性。此外,拟南芥中Polθ缺失会干扰外源DNA片段插入基因[42]。虽然Polθ介导的Alt-EJ修复易出错,但与其他在缺失情况下进行修复并可能导致总基因组畸变的过程相比,Polθ介导的Alt-EJ修复通常是更安全的选择,具有维持基因组稳定性的作用。然而学术界也有不同的结论,Mateos-Gomez等[19]报道Polθ本身会导致基因组不稳定,这可能源于其在Alt-EJ 中的诱变作用。多个研究发现,Polθ损耗可减少染色体易位和紫外线相关突变,其过表达增加DNA 损伤标记物并损害细胞周期进展[4,19,43]。目前,Polθ 对于基因组稳定的影响还存在一定争议,这可能与特定的DSB诱导因素和细胞类型有关。
4 Polθ与肿瘤发生
4.1 Polθ在多种肿瘤组织中高表达
由于Polθ在DNA合成时具有低保真度的特点,当它在不同的时间和位点发挥作用时,其升高可能导致染色体重排,甚至肿瘤的发生。一般情况下,在正常细胞中Polθ 低表达或不表达,仅在骨髓和淋巴系统中高表达。而在恶性肿瘤如结直肠癌、肺癌、胃癌、乳腺癌、卵巢癌和头颈部癌等中,发现Polθ 的高表达[1-4],而且大量临床数据分析发现,Polθ过表达可能与患者预后不良、基因突变、肿瘤分级及无复发生存期缩短有关[1,44]。
4.2 抑制Polθ介导肿瘤细胞合成致死性
在恶性肿瘤细胞中,由于HR修复机制发生缺陷,抑制Polθ 具有显著的合成致死效应[6,19]。Ceccaldi 等[45]发现,在BRCA缺乏的癌细胞中,Polθ 表达升高会增加Alt-EJ 修复活性,以补偿HR缺陷。因此,抑制Polθ 可以作为抗击肿瘤的潜在治疗靶点。目前PARPi 临床多用于治疗HR 缺陷类型肿瘤,但PARPi耐药性问题成为其广泛应用的主要障碍[46]。Li 等[47]发现,Polθ 抑制与PARPi 联合,在HR 缺陷肿瘤中可发挥“双合成致死”作用,即两个或多个基因和两条途径共同导致肿瘤细胞死亡,且敲除POLQ基因,可能会提高HR 缺陷肿瘤细胞对PARPi的敏感性[7]。
4.3 抑制Polθ促进肿瘤细胞死亡的其他机制
研究表明,Polθ也与先天免疫反应激活存在联系。肿瘤细胞中HR 修复不足,抑制Polθ 会导致Alt-EJ修复减少,造成基因组不稳定,进而促进免疫细胞对癌细胞的免疫识别和免疫破坏[48]。其潜在机制为:a. 新抗原生成[49];b. cGAS/STⅠNG 通路激活[50];c. 免疫细胞死亡诱导[51]等,来触发癌细胞的免疫原性。近期,Patterson-Fortin 等[52]也发现,在BRCA缺陷癌症中,抑制Polθ 会激活cGAS/STⅠNG 通路,导致T 细胞浸润和活性增加,并对免疫检查点阻断剂产生敏感性。
5 Polθ小分子抑制剂
靶向癌细胞的Alt-EJ修复通路既可以选择性地杀死肿瘤细胞,同时又不影响正常细胞存活,因此Polθ 是治疗HR 缺陷癌症的一个很有前景的靶点[53],此外,它还可以作为人类癌症免疫肿瘤治疗高应答的潜在生物标志物。现阶段已经报道多个高特异性且有效的Polθ小分子抑制剂用于阻断HR缺乏的各种癌症(表1)。
5.1 ART558、ART812和ART899
Zatreanu 等[54]发现一种有效的、选择性的小分子Polθ-Pol 抑制剂ART558,通过破坏Polθ-Pol DNA 复合物的稳定性降低了生产能力,进而抑制引物的延伸。该研究发现,在BRCA2-/-的结直肠腺癌上皮细胞DLD1、胰导管腺癌肿瘤细胞CAPAN1和视网膜色素上皮细胞RPE 中给与1~10 μmol/L ART558 治疗后,可诱导肿瘤细胞SL[6,54]。在BRCA2-/-DLD1细胞中,产生了DNA损伤相关的生物标志物,包括核γH2AX 病灶、磷酸化H2AX、染色体异常和微核的增加,这可能是导致合成致死的原因。在该细胞中,同时给与ART558 和PARPi奥拉帕尼处理对细胞存活、克隆培养率和凋亡的影响远大于DLD1.BRCA2野生型细胞[54]。在BRCA-/-RPE细胞中,0.5 μmol/L ART558及0.1 μmol/L奥拉帕尼同时处理后,细胞存活显著降低[54]。ART558不仅能使BRCA-/-肿瘤细胞合成致死,还能减弱由p53 结合蛋白1 抗体(p53-binding protein 1,53BP1)缺陷引起的PARPi 耐药性[54]。53BP1-/-和BRCA1-/-的患者源性肿瘤体外培养类器官对PARPi奥拉帕尼或卡铂耐药,但是对ART558 处理敏感,肿瘤在1~100 μmol/L ART558 处理时存活显著减少[54]。Rodriguez-Berriguete 等[55]在结肠癌细胞HCT116、大细胞肺癌细胞H460和膀胱癌细胞T24中发现,1 μmol/L ART558 即可增加3 种肿瘤细胞的放射敏感性。由于ART558 稳定性较差,Zatreanu 等[8]进一步优化衍生出新Polθ-Pol 抑制剂,ART812,发现对BRCA1/SHLD2-/-(SHLD2为在DNA 修复中起关键性作用的一种蛋白质)乳腺癌细胞MDA-MB-436荷瘤大鼠模型给与100 mg/kg ART812 时即有明显的肿瘤抑制作用[54]。随后Rodriguez-Berriguete 等[55]又研制出另一种Polθ-Pol 抑制剂——ART899。1 mmol/L 和3 mmol/L ART899处理HCT116和H460细胞,可明显增加肿瘤细胞放疗敏感性[55]。
5.2 新生霉素(novobiocin,NVB)
NVB 是一种新发现的特异性的Polθ-Hel 抑制剂[7],它与Polθ 的配体蛋白结合阻止Polθ 募集到DNA 损伤部位从而抑制Alt-EJ 修复。NVB 可以剂量依赖性诱导HR 缺陷的肿瘤死亡[7]。Zhou 等[7]发现,在骨肉瘤细胞U2OS 中,分别使用50 μmol/L、100 μmol/L NVB 时,MMEJ 活性下降约50%,而对HR活性没有明显影响。NVB可以与PARPi协同杀死HR缺陷型肿瘤,在BRCA-/-RPE细胞中,100 μmol/L NVB 与10 nmol/L PARPi 鲁卡帕尼同时作用导致细胞提前死亡[7]。HR-/-人源肿瘤异种移植 (patient-derived tumor xenografts,PDXs)肿瘤模型(DF83 和DF59)中,NVB 与PARPi 奥拉帕利同时作用,使PARPi 耐药性减弱,肿瘤完全/提前消退[7]。因此,NVB 和PARPi 联合使用比单独使用PARPi能更有效地杀死HR缺乏的肿瘤,且联合治疗可预防PARPi的耐药性。Polθ表达水平是NVB 敏感性的预测性生物标志物,NVB还可诱导DSB 末端切除和RAD51 病灶增加[7]。Patterson-Fortin等[56]发现,随着C-NHEJ关键蛋白DNA-PK抑制剂培塞替布浓度增加(1~5 μmol/L),给与100 μmol/L NVB,使TP53 缺陷肿瘤细胞,如非小细胞肺癌细胞A549 和H460 细胞毒性逐渐增强,且NVB可以减弱TP53突变肿瘤细胞对培塞替布的耐药性。因此,靶向DNA-PK 抑制剂和Polθ抑制剂组合可能为TP53 突变实体瘤提供一种合理的治疗策略。
5.3 RP-6685
RP-6685 是一种强效针对Polθ-Pol 的口服抑制剂,它对Polθ 表现出极好的选择性,对其他几种DNA 聚合酶(Pols α、ε、γ、λ 和μ) 均没有活性[57]。Bubenik 等[57]发现,在人结肠癌细胞BRCA2-/-HCT116 的小鼠异种肿瘤移植模型中,给与80 mg/kg RP-6685 治疗8 d,肿瘤细胞即被明显抑制,并且出现微核和γH2AX增加的DNA损伤标志物。但遗憾的是,肿瘤细胞对RP-6685会产生耐药。也许该抑制剂与其他化疗药联合使用会产生更好的疗效,相关工作还需进一步探索和研究。因此,靶向Polθ-Pol活性的抑制剂可能成为进一步开发的候选药物。
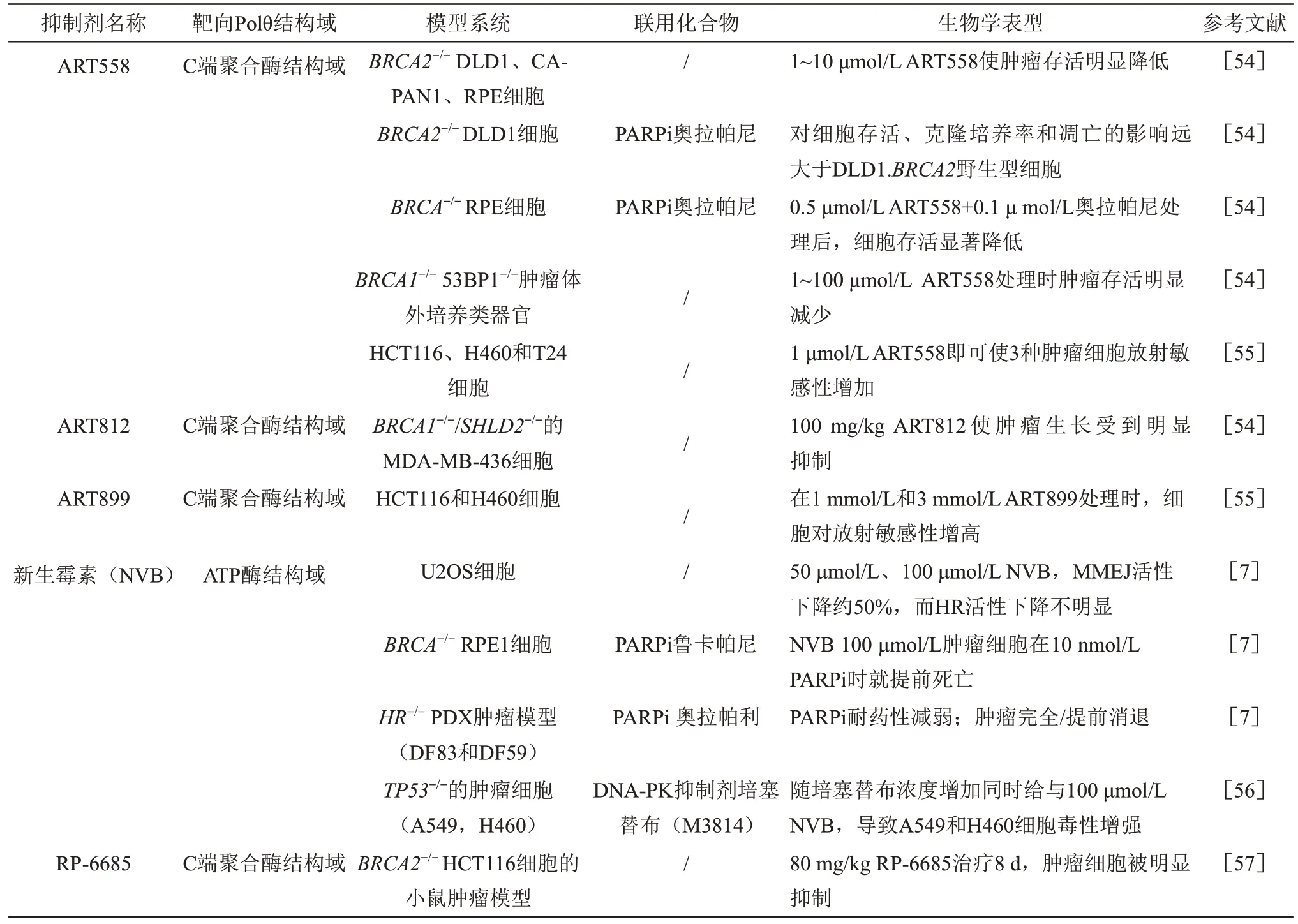
Table 1 Small molecule inhibitors targeting Polθ and tumor lethality表1 Polθ小分子抑制剂与肿瘤致死
6 抑制Polθ有助于提高外源基因靶向整合效率
生理情况下,由于Polθ 与DNA 修复蛋白相互作用,会抑制HR 通路,进而导致依赖HR 通路的外源基因靶向整合效率低下。近期大量研究发现,抑制Polθ 有可能增加HR,可大幅度提高外源基因的靶向效率[6,19](表2)。另一方面,在正常细胞中,由于内在的HR修复机制的存在,抑制Polθ一般不会产生严重合成致死效应。Zelensky 等[58]使用CRⅠSPR/Cas9 技术在小鼠胚胎干细胞(embryonic stem cell,ES)中同时敲除POLQ基因和C-NHEJ关键分子Ku80时,发现HR基因靶向效率在Rad54基因座和Pim1基因座分别提高了23%和3 倍,进而敲除POLQ基因和联用DNA-PK 抑制剂NU-7441 时,外源基因在Rad54基因座的9 号和18 号外显子位置的外源基因靶向整合效率分别提高9%、 11%, 同时发现随机整合(random integration,RⅠ)的降低主要是通过抑制Polθ 介导的Alt-EJ 通路,而抑制C-NHEJ 通路对外源基因敲入效率影响不明显。进一步研究发现,同时抑制Polθ和C-NHEJ关键蛋白Ku70/Ku80/LIG4,抑制RⅠ会更为明显[58-60]。Saito等[60]也发现,在B淋巴细胞白血病细胞中同时敲除POLQ和LIG4基因,基因靶向效率提高约99.4%。同样,在衣藻中使用CRⅠSPR/Cas9 体系敲除POLQ基因,发现HR 修复效率提高了28%~97%[59]。Arai 等[61]在小鼠ES 中利用shRNA 敲低POLQ基因并联合DNA-PK 抑制剂M3814,提高双等位基因敲入效率15倍。另外,NVB 与M3814 联用可协调提高双等位基因敲入效率[61]。另一项研究表明,在LIG4-/-HEK293 细胞中,用RP-6685敲低POLQ基因,靶向整合效率提高了2倍[9]。综上所述,通过敲除或用小分子抑制剂阻断Polθ 介导的Alt-EJ 通路有助于提高HR 通路活性,增加外源基因靶向整合效率。
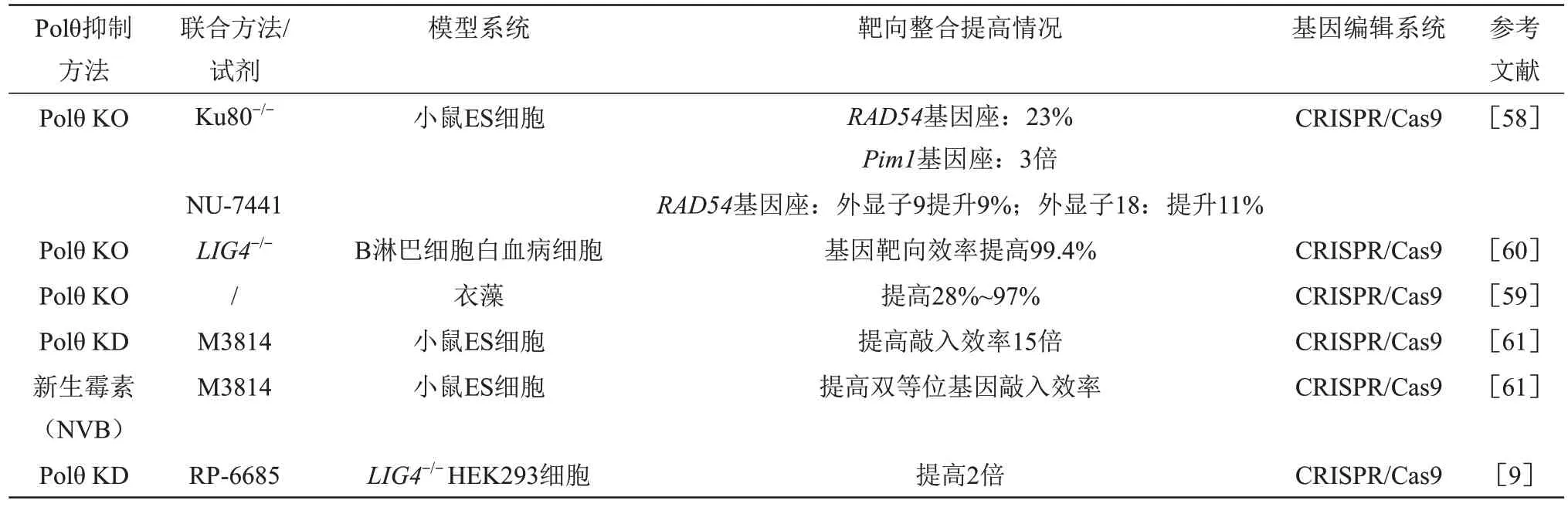
Table 2 Inhibition of Polθ improves target integration efficiency表2 抑制Polθ提高HR效率
7 总结与展望
Polθ是Alt-EJ修复通路的关键分子,生理条件下主要参与维持基因组稳定性,而其异常高表达则会促进多种癌症的发生发展。现阶段,科学家已经研发多种靶向Polθ的小分子抑制剂,发现阻断Alt-EJ修复通路可增加肿瘤细胞对放疗或化疗敏感性,逆转部分肿瘤的PARPi耐药。因此,Polθ有望成为癌症治疗的新靶点,同时也可作为肿瘤免疫治疗高反应的潜在生物标志物。除此之外,抑制Polθ 活性还可以提高外源基因发生同源重组的效率,在基因靶向整合方面具有潜在应用前景。相信随着人们对Polθ介导Alt-EJ通路机制研究的不断深入,越来越多的相关应用会在生物制药领域发挥重要的有益作用。
