膜蛋白ATAD3A在线粒体质量控制中的作用*
2024-03-23尚画雨
张 舵 夏 志 尚画雨**
(1)成都体育学院运动医学与健康学院,成都 610041;2)温州大学体育与健康学院,温州 325035;3)井冈山大学体育学院,吉安 343009)
线粒体参与细胞能量供应的同时会产生活性氧(reactive oxygen species,ROS),其生成增加将诱发线粒体结构损伤、线粒体DNA(mitochondrial DNA,mtDNA)突变和蛋白质折叠紊乱等失稳态现象[1]。线粒体稳态失调易引起细胞功能障碍,从而成为衰老相关疾病的重要发病机制[2]。因此,线粒体被赋予多种质量控制系统,通过调节线粒体形态、功能与质量而维持一定数量功能正常的线粒体以满足细胞代谢需求。线粒体质量控制系统可通过线粒体融合蛋白1/2(mitofusin1/2,Mfn1/2)和发动蛋白相关蛋白1(dynamin-related protein 1,Drpl)依赖线粒体融合分裂以修复或裂解受损线粒体, 并通过PTEN 诱导激酶(PTEN-induced putative kinase protein 1,PⅠNK1)介导线粒体自噬以清除损伤线粒体,还能通过线粒体生物合成而产生新的线粒体,共同维持线粒体稳态[3-4]。
线粒体内膜蛋白三磷酸腺苷酶家族蛋白3(ATPase family AAA domain-containing protein 3,ATAD3)最初发现于小鼠肝细胞线粒体膜[5],其家族成员ATAD3A 具有特殊而复杂的结构,与线粒体质量控制密切相关,参与多个关键的生理和病理过程。例如,ATAD3A 参与维持线粒体嵴形态[6]、内膜结构和呼吸功能[7]。再如,它可与Drp1 相互作用,通过形成寡聚体而正性调节Drp1依赖性线粒体分裂,导致亨廷顿病患者多功能干细胞内线粒体断裂及mtDNA 缺失[8]。又如,它可促使PⅠNK1 向线粒体输入以抑制PⅠNK1 在线粒体外膜上高表达和介导线粒体自噬,从而提升造血干细胞存活率,维持造血稳态[9]。基于此,本文对近年来ATAD3A 蛋白的相关研究进行梳理,并归纳其在线粒体质量控制方面的调控作用,以期为ATAD3A相关疾病(神经退行性疾病和癌症等)的新药研发提供参考资料。
1 ATAD3A
ATAD3A 被认为是ATAD3 家族最早的起源蛋白质,由586个氨基酸组成,在胚胎期至成年阶段的所有组织中广泛表达,尤其是中枢神经系统中最为丰富。ATAD3A蛋白高度保守,其N端定位于线粒体内膜且与外膜相互作用,参与调节蛋白质相互作用和ATAD3A寡聚体的形成,包含1个富含脯氨酸残基的灵活区域(proline-rich motif,PRM)、2个跨膜结构域(trans-membrane domains,TM,包含TM1 与TM2 子域) 和2 个卷曲螺旋结构域(coiled-coil domains,CC,包含CC1与CC2子域),其中CC结构域参与正性调节Drp1依赖性线粒体分裂[8]。ATAD3A C 端的ATP 酶结构域(AAA+ATPase domains,AAA+ATPase) 位于线粒体基质[10],参与负性调节Drp1介导的线粒体分裂,且该结构域包含的Walker A 和Walker B 子域亦分别参与调节ATP的结合和水解[11-12](图1)。
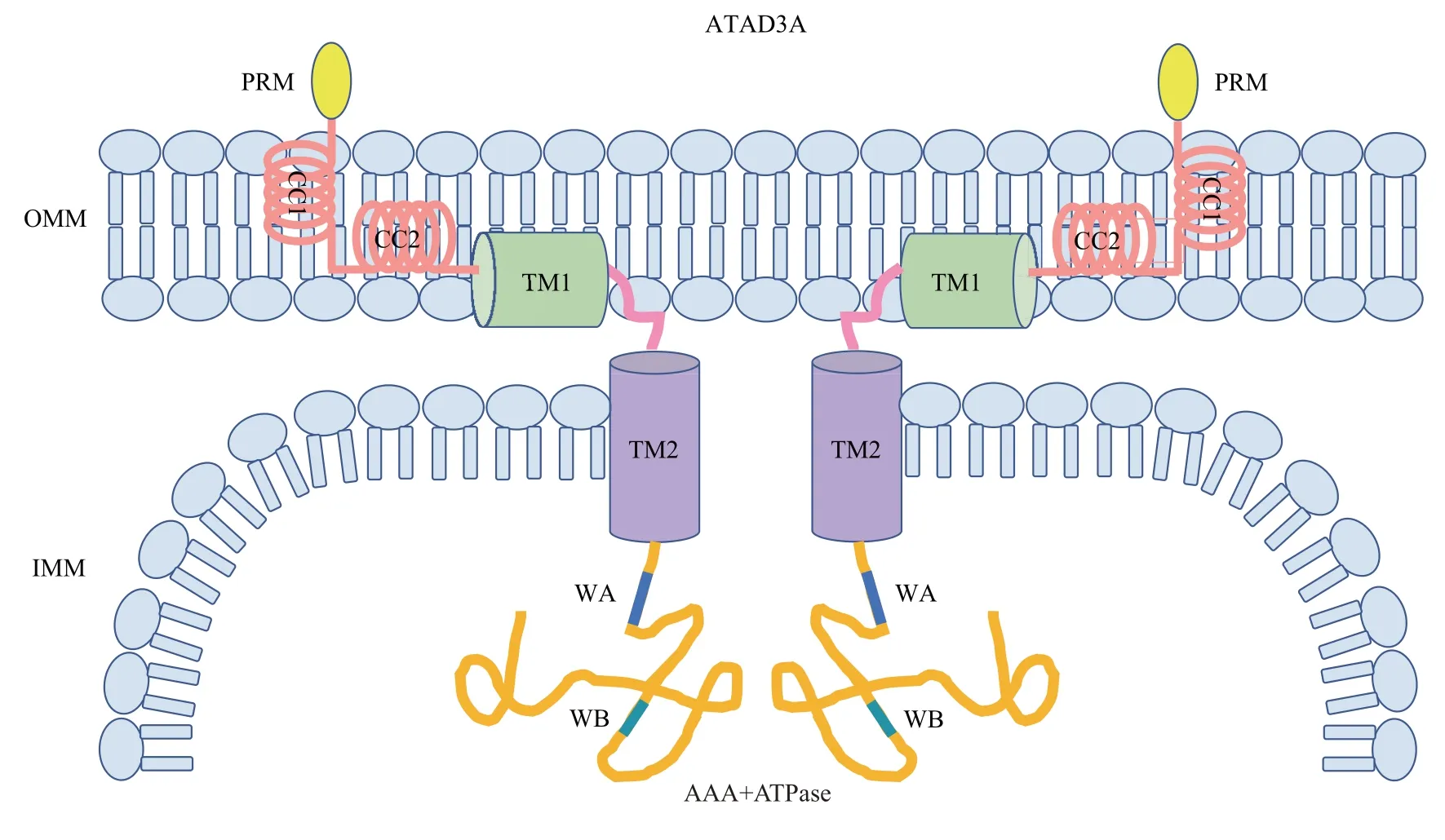
Fig. 1 Diagram of protein structure of ATAD3A图1 ATAD3A蛋白结构图
目前已知ATAD3A 是一个多结构域蛋白质,从而可与线粒体嵴结构蛋白60(MⅠCOS subunit/Mitofilin, Mic60)[6]、 线粒体转录因子A(mitochondrial transcription factor A,TFAM)[13]、Drp1[8,14]、自噬/苄氯素1 调节因子1(autophagy/beclin-1 regulator 1,AMBRA1)[15]、分子伴侣受体蛋白(sigma-1 receptor,sig-1R)[16]、PⅠNK1[9,17]、脂质激酶(acylglycerol kinase,AGK)[18]、细胞色素C 氧化酶亚基56[19]、范可尼贫血互补群D2[20]、亮氨酸拉链EF-hand 结构域跨膜蛋白1[7]、NⅠMA相关激酶10[21]、神经胶质成熟因子γ[22]、S100 钙结合蛋白B[23]、黏蛋白1[24]、视神经萎缩蛋白1(optic atrophy 1,OPA1)[25]、脂滴包被蛋白2(perilipin 2,PLⅠN2)[26]、电压依赖性阴离子通道蛋白 1 (voltage dependent anion channel 1,VDAC1)[27]等多种蛋白质之间产生相互作用。研究表明,ATAD3A还涉及多方面调节机制:可通过负性调节凋亡诱导因子由线粒体向胞核转位,从而抑制细胞凋亡[25];可通过正性调节哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)-固醇调节元件结合蛋白1c/细胞周期蛋白D1 信号通路,从而促进牛乳生物合成和乳腺上皮细胞增殖[28];亦可与家蚕核型多角体病毒(Bombyx mori nucleopolyhedrovirus,BmNPV)晚期表达因子(late expression factor 11,LEF-11)相互作用,从而促进BmNPV病毒复制[29];还可在线粒体外膜蛋白VDAC1 依赖的情况下与细胞外信号调节激酶 1/2 (extracellular regulated protein kinases,ERK1/2)相互作用以上调线粒体ERK1/2磷酸化表达,从而促进头颈鳞状细胞癌细胞增殖[27]。
2 ATAD3A参与调节线粒体质量控制
2.1 ATAD3A参与调节线粒体结构
线粒体嵴是线粒体内电子传递和氧化磷酸化的主要场所,其形态变化与线粒体功能密切相关,从而直接影响细胞能量代谢。研究表明,ATAD3A可通过与Mic60等嵴结构蛋白相互作用而调节线粒体嵴结构。Hu 等[6]在ATAD3A基因敲除(ATAD3Aknock-out,ATAD3AKO)的HeLa 细胞内观察到嵴断裂、变短且嵴之间相互融合致使其数量明显减少,嵴形态调控蛋白Mic60、OPA1 和YME1L(YME1 like 1 ATPase)蛋白表达显著降低,且免疫共沉淀实验结果显示ATAD3A 可与Mic60 相互作用,表明ATAD3A可能与Mic60等协同调节线粒体嵴的形态。
Peralta 等[30]报道指出,骨骼肌特异性ATAD3AKO 小鼠在出生两个月后体重明显减轻,诱发肌肉质量、力量及功能减退的肌病,表现为骨骼肌线粒体嵴断裂、变短且与线粒体膜呈同心圆排列,线粒体和嵴表面积均明显变小,线粒体嵴形态调控蛋白Mic60与OPA1、呼吸链复合体Ⅰ亚基泛醌氧化还原酶亚基 B8 (NADH ubiquinone oxidoreductase subunit B8,NDUFB8)和复合体V亚基ATP 合成酶亚基α (ATP synthase subunit alpha,ATP5A)蛋白表达以及mtDNA 拷贝数、胆固醇转运至线粒体的水平(小鼠腓肠肌胆固醇酯/血浆游离胆固醇比值)均显著降低。以上结果提示,ATAD3A可能参与维持骨骼肌线粒体嵴结构稳态以保障线粒体功能的正常运行,从而在骨骼肌质量控制中发挥重要作用。随后,Peralta 等[31]在ATAD3A基因突变诱发的新生儿小脑发育不全患者成纤维细胞中亦证实线粒体嵴结构受损,同时ATAD3A蛋白质表达明显下调,而骨骼肌线粒体呼吸链复合体Ⅰ~V 亚基的蛋白质表达均无明显变化,这与其在骨骼肌特异性ATAD3AKO 小鼠中的研究结果[30]不一致,他们推测线粒体嵴被破坏时呼吸链复合体的稳定性可能存在物种或组织差异。上述研究结果表明,线粒体内膜蛋白ATAD3A 是治疗骨骼肌病变和新生儿小脑发育不全的潜在靶点,其参与维持线粒体嵴结构稳态可能是通过与Mic60相互作用而实现,但ATAD3A基因缺失致使线粒体变小和嵴损伤的具体机制仍有待进一步研究阐明。
2.2 ATAD3A参与调节线粒体功能
研究证实,ATAD3A作为机体能量代谢所必需的组分,是调节线粒体功能的关键膜蛋白。最初Hoffmann等[32]观察到,ATAD3基因敲除的秀丽隐杆线虫呈现出胚胎死亡、幼虫发育停滞和性腺功能障碍等异常现象,同时体壁肌线粒体呼吸链复合体Ⅰ和柠檬酸合成酶活性以及肠内脂肪含量均明显降低,表明ATAD3 参与维持秀丽隐杆线虫正常的线粒体功能和生长发育。Lee等[33]近期的报道指出,小鼠原代颅盖骨成骨细胞中缺乏ATAD3A 将导致脂滴含量和ROS 水平明显上调,线粒体嵴密度、膜电位、mtDNA拷贝数和ATP生成量均显著下调,而ATAD3A 过表达则可有效逆转上述反应。鉴于成骨细胞分化和成熟与其线粒体功能正性相关[34],上述结果表明,ATAD3A可能通过维持线粒体稳态以正性调节成骨细胞分化与成熟。Arguello等[7]在小鼠神经元中特异性敲除ATAD3A 后发现,小鼠存活率和运动协调能力下降,神经元线粒体明显变小且嵴断裂,呼吸链复合体V 亚基ATP5A 蛋白表达、mtDNA 和磷脂酰胆碱含量均明显减少,提示ATAD3A亦参与维持神经元线粒体结构与功能,从而对神经元细胞存活发挥关键作用,但具体机制仍有待后续研究予以确认。
此外,Cooper等[35]报告了一例由ATAD3A突变导致遗传性痉挛性截瘫的患者,其皮肤成纤维细胞线粒体破碎并被明显拉长,呼吸链复合体Ⅱ亚基琥珀酸脱氢酶复合铁硫亚基B (succinate dehydrogenase complex iron sulfur subunit B,SDHB)、复合体Ⅳ亚基细胞色素C 氧化酶1(mitochondrially encoded cytochrome C oxidase 1,MTCO1)、Drp1 及mTOR 下游p-S6 蛋白表达均显著下调且p-mTOR蛋白表达亦呈现下降趋势,同时溶酶体数量明显增加,提示ATAD3A 突变除引起线粒体氧化磷酸化功能障碍外,还可能通过抑制mTOR活化而促进溶酶体生成。Dorison等[36]近期亦观察到,一对由ATAD3A 突变引起的轴突感觉运动神经病变并伴有新生儿白内障的兄妹患者皮肤成纤维细胞中线粒体呼吸链复合体Ⅰ亚基NDUFB8和复合体Ⅳ亚基MTCO2蛋白表达均显著下调,且骨骼肌线粒体肿胀、嵴排列紊乱。上述研究结果表明,ATAD3A 参与调节线粒体正常形态和呼吸功能,这可能是其维持线粒体完整性以及细胞分化与成熟的关键所在。未来可进一步开展家族遗传性神经系统疾病研究,以明确ATAD3A 在遗传性神经病变中的作用机理及其与神经功能之间的关系。
2.3 ATAD3A参与调节线粒体动力学
线粒体是高度动态的细胞器,在细胞内不断分裂、融合,有利于线粒体实现物质交换和能量供应,调节线粒体形态、功能、自噬及细胞凋亡。线粒体分裂主要调控蛋白为Drp1,定位于胞质中,可被线粒体外膜上的受体蛋白线粒体分裂因子、49/51 ku线粒体动力蛋白和Drp1募集至外膜以介导线粒体分裂。此外,线粒体融合则由线粒体外膜融合蛋白Mfn1和Mfn2及内膜蛋白OPA1介导[37]。目前研究结果表明,ATAD3A 参与正性/负性调节线粒体分裂与融合。
一方面,ATAD3A 可抑制Drp1 介导的线粒体分裂。Fang 等[10]最初观察到肺腺癌细胞(H23、H226、 H838、 H1437、 H2009、 H2087、 A549、SK-MES-1)中ATAD3A mRNA 和蛋白质表达明显上调,且其顺铂敏感度随ATAD3A 蛋白表达上调而显著下降,而在肺腺癌细胞A549和H838中敲除ATAD3A后发现,线粒体碎片化程度和顺铂敏感度均明显升高,故推测ATAD3A 可能参与抑制线粒体分裂和肺腺癌细胞凋亡,但其抗凋亡活性和调节线粒体碎片化之间的联系尚不明确。Gilquin 等[11]在ATAD3A的C端AAA+ATPase结构域突变的胶质瘤U373 细胞中观察到线粒体分裂明显增加,而敲低Drp1 后线粒体分裂被抑制,表明ATAD3A 的AAA+ATPase 结构域参与负性调节Drp1 介导的线粒体分裂,但ATAD3A 对线粒体分裂的抑制作用是否影响U373癌细胞活性仍有待研究进一步确认。
另一方面,ATAD3A 亦能促进Drp1 依赖性线粒体分裂。最近Zhao 等[8]在人胚肾HEK293 和HeLa细胞内敲除ATAD3A的CC结构域后,线粒体明显趋向拉长和融合,且线粒体Drp1 蛋白表达显著下调,从而提示ATAD3A的CC结构域可能通过介导Drp1 线粒体转位而促进线粒体分裂。此外,在介导线粒体分裂过程中,Drp1 形成螺旋寡聚体包裹住线粒体外膜并诱导其分裂[38],而亨廷顿病的发生发展与Drp1 过度激活形成寡聚体所致线粒体过度分裂密切相关[39]。Zhao等[8]在亨廷顿病患者多能干细胞中观察到ATAD3A 寡聚体明显增多且与Drp1 存在相互作用,而在亨廷顿病小鼠纹状体HdhQ7 和HdhQ111 细胞中敲除ATAD3A 后,Drp1寡聚体数量和线粒体Drp1蛋白表达明显下调。选取短肽抑制剂DA1 处理亨廷顿病小鼠纹状体HdhQ111 细胞,结果显示,DA1 可与ATAD3A 相互作用,同时ATAD3A、Drp1 寡聚体的形成、线粒体Drp1 蛋白表达及其分裂程度明显降低,然而DA1 对Drp1 寡聚体形成的抑制作用在ATAD3A 敲低后被消除,表明DA1 抑制Drp1 活化需要ATAD3A的参与。以上结果提示,短肽抑制剂DA1可能通过与ATAD3A 相结合,阻止ATAD3A 与Drp1 相互作用,进而减少ATAD3A 寡聚体数量以及线粒体Drp1 蛋白表达,最终抑制亨廷顿病模型下线粒体过度分裂。因此,ATAD3A有望成为治疗亨廷顿病的关键因子。
另外,Watanabe 等[16]采用免疫共沉淀技术在HeLa 细胞内观察到线粒体相关内质网膜上的分子伴侣蛋白sig-1R与ATAD3A存在相互作用,且在小鼠运动神经元与HeLa 细胞内通过给予sig-1R 敲除或过表达处理,可分别上调和下调ATAD3A 寡聚体数量及线粒体碎片化程度。然而,在ATAD3A敲低的HeLa 细胞中再次敲低或过表达sig-1R 后发现,线粒体形态均无明显变化。此外,他们还在肌萎缩侧索硬化症小鼠的脊髓运动神经元中观察到sig-1R与ATAD3A共定位的量和ATAD3A蛋白表达显著减少,而ATAD3A 寡聚体数量和线粒体碎片化程度明显增加。如前所述,ATAD3A寡聚体可介导Drp1 依赖性线粒体分裂[8],上述结果表明,sig-1R可通过与ATAD3A相互作用以抑制ATAD3A寡聚体的形成,从而阻止线粒体分裂,故sig-1R和/或ATAD3A 的靶向促进可能是由线粒体异常分裂所致肌萎缩侧索硬化症治疗的有效方法,但其具体作用及相关机制仍有待深入研究。
黄琴[14]构建阻塞性睡眠呼吸暂停综合征大鼠模型,成模大鼠呈现出烦躁不安、嗜睡、呼吸加深加快等病症,其探索水迷宫平台时间、海马中Drp1Ser616磷酸化表达和神经元细胞凋亡明显增加,而Mfn1 和Mfn2 蛋白表达显著减少。此外还发现,海马特异性敲低ATAD3A 的成模大鼠探索水迷宫平台时间、Drp1Ser616磷酸化表达明显减少,而穿越平台次数、Mfn1和Mfn2蛋白含量则显著增多。以上结果提示,阻塞性睡眠呼吸暂停综合征大鼠认知障碍可能是由于线粒体高度分裂所致,而干扰海马ATAD3A表达可有效抑制线粒体分裂并促进受损线粒体的融合修复,从而改善阻塞性睡眠呼吸暂停综合征大鼠认知障碍,同时亦再次表明ATAD3A 是治疗神经系统疾病的线粒体潜在靶点。
总体而言,线粒体内膜蛋白ATAD3A 参与负性调节线粒体融合,其在线粒体分裂中的作用可能具有细胞类型特异性,但其具体作用(促进或抑制)迄今仍不明确。因此,深入研究ATAD3A 在癌细胞(如肺腺癌与胶质瘤)凋亡以及线粒体分裂/融合失衡引起的神经系统疾病(如亨廷顿病、肌萎缩侧索硬化症和阻塞性睡眠呼吸暂停综合征认知障碍)中的调控机制可能具有重要意义。
另有报道指出,线粒体拟核是由mtDNA 与拟核相关蛋白(如TFAM)共同形成的核蛋白复合物,锚定于线粒体内膜[40]。近期Ⅰshihara 等[13,41]通过免疫共沉淀试验在HeLa 细胞内观察到ATAD3A的AAA+ATPase结构域与TFAM存在相互作用。另外,在Drp1敲低的HeLa细胞内发现线粒体趋向融合、拟核聚集在线粒体内膜,同时线粒体呼吸链复合体Ⅰ亚基NDUFB8、复合体Ⅱ亚基SDHB、复合体Ⅳ亚基MTCO1蛋白表达明显降低,复合体ⅠⅠⅠ亚基泛醇-细胞色素C 还原酶核心蛋白2(ubiquinol-cytochrome C reductase core protein 2,UQCRC2)蛋白表达亦呈现下降趋势,然而在HeLa细胞内同时敲低Drp1与ATAD3A则可有效增加线粒体呼吸链复合体亚基NDUFB8、UQCRC2、MTCO1 蛋白表达并减少拟核聚集。此外,在单独敲低ATAD3A 的HeLa 细胞内观察到线粒体拟核变小、数量增多,且移动速度显著减慢,而mtDNA拷贝数无明显变化。他们推测,在线粒体分裂受阻的情况下线粒体呼吸功能受损,此时拟核可能在线粒体内快速运动而逐渐变大并聚集,然而ATAD3A的靶向抑制则可有效提升线粒体呼吸功能并降低拟核移动速度,导致拟核变小且聚集减少。但是,迄今尚未明确ATAD3A 作为TFAM 相互作用蛋白是否参与调控mtDNA 质量控制,亦不清楚其在线粒体呼吸功能、融合分裂以及拟核形态、分布变化等过程中的不同调节作用究竟如何整合至协调反应之中。
2.4 ATAD3A参与调节线粒体自噬
线粒体自噬作为线粒体质量控制的重要机制之一,能够及时降解清除细胞内受损/多余线粒体以维持细胞正常生命活动。线粒体自噬由多种途径介导,先前研究表明,帕金森病相关蛋白PⅠNK1 及其下游蛋白Parkin(Parkinson protein 2)协同介导哺乳动物细胞线粒体自噬:在正常线粒体中PⅠNK1会被一些蛋白酶体所降解,当线粒体损伤时,蛋白酶体催化作用受阻,阻滞了PⅠNK1 向内膜转运,使PⅠNK1 大量聚集在线粒体外膜,随即磷酸化Parkin并将其招募到受损线粒体上,后者进而介导线粒体自噬以清除损伤线粒体[42-43]。近期研究证实,ATAD3A 作为线粒体膜蛋白参与调节PⅠNK1-Parkin 介导的线粒体自噬。Jin 等[9]报道指出,与野生型小鼠相比,骨髓造血细胞ATAD3AKO 的小鼠死亡率明显升高,其祖细胞和造血干细胞数量显著减少,同时线粒体中PⅠNK1、Parkin、泛素(ubiquitin,Ub) 和微管相关蛋白1 轻链3-Ⅱ(microtubule-associated protein 1 light chain 3-Ⅱ,LC3-Ⅱ) 蛋白表达以及线粒体外膜转位酶20(translocase of the outer mitochondrial membrane 20,TOM20)与溶酶体相关膜蛋白2共定位的量均明显增加,提示ATAD3A 负性调节PⅠNK1-Parkin 介导的线粒体自噬。鉴于PⅠNK1 通常依赖于TOM 复合物和线粒体内膜转位酶(translocase of the inner mitochondrial membrane,TⅠM)复合物以导入线粒体内膜而被裂解[44-45],Jin 等[9]在ATAD3AKO 的小鼠骨髓细胞中还观察到PⅠNK1 与TOM40、TⅠM23 的相互作用均被显著抑制,提示ATAD3A是PⅠNK1 输入线粒体所必需的:ATAD3A 作为TOM 和TⅠM 复合物之间的桥接因子,促进PⅠNK1从线粒体外膜转运至内膜,再经线粒体加工蛋白酶和内膜菱形蛋白酶依次切割而被降解,从而抑制PⅠNK1在线粒体外膜上累积及其对胞质Parkin的招募,最终阻止线粒体自噬。值得注意的是,在ATAD3AKO 的小鼠骨髓造血细胞内进一步敲除PⅠNK1后线粒体自噬水平显著下调,同时祖细胞和造血干细胞数量明显增加,这表明ATAD3A 可能通过抑制PⅠNK1 依赖性线粒体自噬以维持造血稳态。未来可进一步开展骨髓造血细胞功能的离体研究,以明确ATAD3A 在正性调节造血稳态中的作用机理以及PⅠNK1 介导的线粒体自噬与造血细胞功能之间的关系。
另有研究表明,AMBRA1 是Unc-51 样自噬活化激酶1 和beclin-1 依赖性细胞自噬[46]以及PⅠNK1-Parkin 依赖性线粒体自噬的正性调控因子[47],并可作为受体蛋白介导PⅠNK1-Parkin 非依赖性线粒体自噬[48]。近期Di Rienzo 等[15]在线粒体解偶联剂羰基氰化物氯苯腙处理过的SH-SY5Y和HeLa 细胞中观察到,AMBRA1 分别与PⅠNK1、TOM20 及ATAD3A 存在相互作用,且使用siRNA干扰AMBRA1 表达后SH-SY5Y 和HeLa 细胞内LC3-Ⅱ/LC3-Ⅰ比值显著升高,线粒体PⅠNK1 和p-UbSer65蛋白表达明显降低,在此基础上沉默ATAD3A 基因表达反而显著上调AMBRA1 和线粒体PⅠNK1蛋白表达。由此可见,ATAD3A既可在线粒体内外膜之间介导PⅠNK1的线粒体输入与降解,又能与自噬受体AMBRA1 相互作用并下调AMBRA1 及线粒体PⅠNK1 蛋白表达,从而负性调节PⅠNK1 介导的线粒体自噬,以上研究结果均提示了ATAD3A 参与线粒体自噬途径的可能机制,但是ATAD3A 能否抑制AMBRA1 介导的线粒体自噬尚需进一步研究确认。
缺氧是实体瘤生长的重要条件,而缺氧诱导因子1α(hypoxia-inducible factor-1α,HⅠF-1α)可介导肿瘤细胞的血管生成以适应缺氧环境,从而促进癌细胞存活、增殖和侵袭[49]。晚期肝细胞性肝癌一线治疗药物索拉非尼可通过下调缺氧诱导因子HⅠF-1α 的蛋白质表达,抵抗肿瘤血管生成,从而抑制肝细胞性肝癌的进展[50]。Wu 等[51]在建立缺氧诱导的索拉非尼抗性肝细胞性肝癌细胞株(Huh7-h-SR和LM3-h-SR)后观察到细胞内HⅠF-1α和耐药相关基因ABCB1、ABCG2基因表达以及线粒体PⅠNK1、Parkin、LC3-Ⅱ蛋白表达均明显上调,同时ATAD3A的mRNA和蛋白质表达均显著下调,而敲低PⅠNK1的Huh7-h-SR细胞对索拉非尼敏感性及凋亡指数明显升高。此外,在ATAD3AKO 的肝细胞性肝癌细胞内同样观察到线粒体PⅠNK1、Parkin和LC3-Ⅱ蛋白表达明显增加,但细胞对索拉非尼敏感性以及凋亡指数明显降低。因此,他们推测ATAD3A 是缺氧诱导肝细胞性肝癌细胞线粒体自噬信号转导的关键介质:缺氧诱导ATAD3A 蛋白表达下调,进而引起PⅠNK1 在线粒体累积及其介导自噬激活,致使肝细胞性肝癌细胞对索拉非尼的敏感性降低。由此可见,ATAD3A可能是对抗索拉非尼耐药的有效靶点,但其具体作用及相关机制仍有待深入研究。
与此类似,吴齐[18]观察到,T 淋巴细胞特异性敲除ATAD3A的B16-F10黑色素瘤荷瘤小鼠胸腺明显缩小,胸腺细胞总数、CD4+、CD8+和双阳性T细胞显著减少,且肿瘤体积和重量及双阴性T细胞明显增加。此外还发现,ATAD3A 和脂质激酶AGK 在T 细胞各亚群中mRNA 表达变化趋势具有高度相似性,而在ATAD3AKO 的荷瘤小鼠T 细胞和AGK 敲低的细胞系中PⅠNK1 蛋白表达均明显增加且线粒体数量显著减少,同时AGK 可上调ATAD3A 磷酸化水平,故推测AGK 可能通过磷酸化ATAD3A 而抑制PⅠNK1 相关的线粒体自噬,从而维持T 细胞的正常发育和抗肿瘤免疫功能,但AGK 磷酸化ATAD3A 的作用机制有待进一步研究确认。上述Wu 等[51]和吴齐[18]的研究结果表明,ATAD3A可抑制PⅠNK1依赖性线粒体自噬,且自噬可能是癌细胞的一种保护机制。
另外,程序性死亡配体1(programmed death ligand 1,PD-L1)可在癌细胞膜上积累,进而通过与T 细胞表面的免疫抑制因子程序性死亡受体1(programmed death-1,PD-1)相结合以促进肿瘤免疫逃逸[52],故临床上常选取PD-1/PD-L1抗体联合一线化疗药物紫杉醇治疗以提升疗效,但仍存在大部分患者对该疗法敏感性低的情况,因而探究影响联合治疗效果的因素是亟待解决的关键问题[53]。最近Xie 等[17]在PD-1/PD-L1 抗体联合紫杉醇化疗无应答患者中,以及经紫杉醇处理的人三阴性乳腺癌BT549 和MB231 细胞内,均观察到受损线粒体(肿胀、嵴断裂)数量和ATAD3A 蛋白表达明显增加,PⅠNK1、线粒体PD-L1蛋白表达以及PD-L1与溶酶体相关膜蛋白1 在线粒体上的共定位显著减少,且联合治疗无应答患者无进展生存期和总生存期明显缩短。此外,在BT549 癌细胞内敲低ATAD3A后发现,浸润性CD8+T细胞数量、PⅠNK1和线粒体PD-L1蛋白表达明显上调,肿瘤生长速率显著下调,而再次恢复ATAD3A 表达则可有效逆转上述反应。Xie 等[17]还观察到,对ATAD3A 敲低的BT549 和MB231 细胞施加蛋白合成抑制剂放线菌酮和自噬抑制剂巴佛洛霉素A1 处理可分别下调和上调线粒体PD-L1 蛋白表达,同时在人胚肾HEK293细胞中发现PⅠNK1可与PD-L1发生相互作用并正性调控线粒体PD-L1蛋白表达。上述结果表明,ATAD3A与PⅠNK1协同介导PD-L1的线粒体转位和降解,且ATAD3A的靶向抑制能促进PD-L1的线粒体易位和PⅠNK1 依赖性线粒体自噬,这可能是提升PD-1/PD-L1 抗体联合紫杉醇化疗效果的有效方法。
上述研究结果表明,多种癌症的发生发展均伴随着ATAD3A基因和/或蛋白质表达的上调[10,17]或下调[18],ATAD3A 不仅是癌症发生[10,17-18,27]和预后[17]机制中不可或缺的关键环节,还可通过与AMBRA1、PⅠNK1、AGK 等因子相互作用,抑制自噬和(或)正性/负性调控多种化疗药物(如索拉非尼、紫杉醇、顺铂等)诱导的自噬或凋亡过程,从而促进或抑制癌细胞存活、增殖和侵袭[9-10,15,17-18]。由此可见,ATAD3A 可能是维持癌细胞自噬和凋亡之间平衡的调节子:一方面,线粒体自噬可能是癌细胞的一种保护机制,可降低化疗药物(如紫杉醇和顺铂)敏感性,导致癌细胞耐药,因而ATAD3A 抑制PⅠNK1 依赖性线粒体自噬对癌细胞可能具有保护作用,是癌细胞耐药的潜在机制[10,17,51]。另一方面,ATAD3A 亦可通过抑制PⅠNK1介导的线粒体自噬以增强癌细胞对索拉非尼的敏感性,从而发挥抗癌作用[51]。ATAD3A 的靶向促进或抑制可能是调控癌细胞化疗药物敏感性的有效方法,但其具体作用及相关机制仍有待深入研究。
游离胆固醇(free cholesterol,FC)过载会导致细胞内线粒体损伤,是非酒精性脂肪性肝病进展的关键因素[54]。最近Chen 等[26]利用胆固醇和酯酰辅酶A-胆固醇酯酰转移酶抑制剂58035诱导人肝癌Huh7 细胞体外脂质积累模型,在此基础上敲除ATAD3A 后细胞内FC 和甘油三酯(triglyceride,TG)水平、脂解负调节因子PLⅠN2 以及自噬相关蛋白COX Ⅳ、 TOM20、 p62/sequestosome-1、LC3-Ⅱ蛋白表达均明显上调,而PⅠNK1蛋白表达显著下调,同时自噬体明显增多。相反,细胞内过表达ATAD3A 则引起p62 蛋白表达显著降低,LC3-Ⅱ蛋白表达无明显变化。由此可见,在FC 过载的肝癌细胞中,ATAD3A缺失可能一方面通过减少自噬流以阻止PⅠNK1 介导自噬体降解,致使受损线粒体堆积,另一方面通过抑制脂解而加重FC 和TG蓄积,从而诱导肝癌细胞功能障碍。以上离体实验结果提示靶向激活ATAD3A 可能有助于预防非酒精性脂肪性肝病的发生发展。然而,Chen等[26]的在体实验并未证实这一观点,他们在非酒精性脂肪性肝病患者和非酒精性脂肪性肝炎大鼠肝脏组织中均观察到ATAD3A 蛋白表达显著上调,推测肝脂质代谢可能涉及到不同亚型肝细胞(如肝窦内皮细胞、肝星状细胞和枯否细胞)之间的相互作用,故目前仍需进一步开展在体研究以验证上述离体实验结果并探明ATAD3A 促进脂解和线粒体自噬之间的关系。
总体而言,ATAD3A 参与正性/负性调控PⅠNK1-Parkin介导的线粒体自噬,发挥促进或抑制癌细胞存活、增殖和侵袭以及介导脂解的作用,在正性调节造血功能稳态方面亦有重要贡献。在后续研究过程中,还需考虑采用siRNA 干扰和/或敲除ATAD3A 基因以探明其是否参与调控药物和/或运动诱导的线粒体自噬,并通过大规模随机试验对癌症/非酒精性脂肪性肝病被试实施标准化药物和/或运动干预,从线粒体质量控制(自噬、融合/分裂)角度探究可能的影响机制。综上所述,本文将ATAD3A与其相互作用蛋白的关系及其对线粒体质量控制的作用归纳于表1。

Table 1 The relationship between ATAD3A and interacting proteins and its regulatory role in mitochondrial quality control表1 ATAD3A与其相互作用蛋白的关系及其在线粒体质量控制中的作用
3 总结与展望
作为一种多功能蛋白,ATAD3A在多种信号通路中均不可或缺。通过与Drp1、Mic60、TFAM、Sig-1R、PⅠNK1、AMBRA1 及AGK 等因子相互作用,它不仅具有促凋亡或抗凋亡活性,且参与维持线粒体嵴形态和氧化磷酸化功能,并可促进或抑制线粒体分裂及PⅠNK1 依赖性线粒体自噬,在负性调节线粒体融合中亦有重要贡献(图2)。但是,目前学界对于该蛋白质的研究仍有待继续深入,后续应考虑在体内或体外不同应激状态下,或在神经退行性疾病及肿瘤等不同模型中,深入研究ATAD3A 与其他相关通路以及调节因子的相互作用,以期探明ATAD3A在调节凋亡信号、mTOR信号和线粒体质量控制等过程中的不同作用究竟如何整合至协调反应之中。尽管ATAD3A 发挥上述功能的分子机制尚有待未来研究进一步阐明,但这一特殊的ATAD3 家族蛋白可能成为药物和/或运动干预等诸多独立生物进程中的共同靶点。
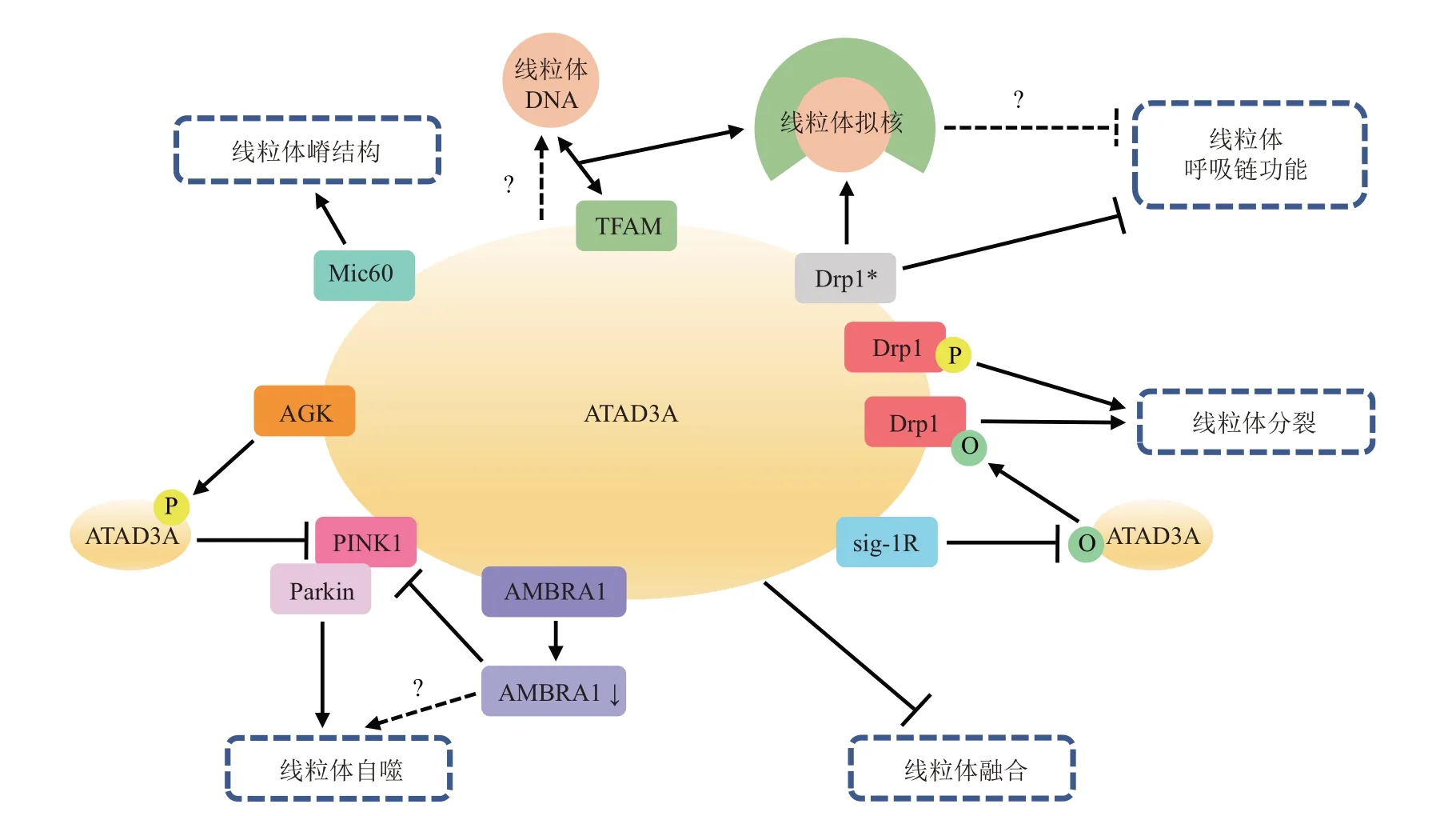
Fig. 2 Network diagram of ATAD3A interacting proteins involved in regulating mitochondrial quality control图2 参与调节线粒体质量控制的ATAD3A相互作用蛋白网络图
