磷脂酰肌醇3激酶通过Akt和ERK抑制盘基网柄菌细胞的趋电性*
2024-03-23葛晓雪蒋锐达王晓燕高润池
葛晓雪 蒋锐达 王晓燕 高 晶 高润池
(1)云南师范大学生命科学学院,昆明 650500;2)云南大学农学院,昆明 650500;3)生物能源持续开发利用教育部工程研究中心,昆明 650500)
细胞定向迁移是多细胞有机体完成正常生命活动的前提和基础,也是发生机体病变的一个重要因素。环境中的许多信号都会影响细胞的迁移方向,如化学因子指导细胞发生的趋化性运动、生物电信号指导细胞发生的趋电性运动等。目前,人们对细胞响应胞外化学因子而发生定向迁移的分子机制研究的比较透彻,而对细胞响应环境中生物电场的分子机制则并不十分清楚。
在细胞的趋化性机制研究中,人们利用模式生物盘基网柄菌(Dicyostelium discoideum)证实了磷脂酰肌醇3 激酶(PⅠ3Ks)的调控作用。研究发现,盘基网柄菌中的PⅠ3K1 和PⅠ3K2 都具有Ras 结合结构域,并且都是趋化试剂刺激PⅠ3K 活化所必须的[1]。在趋化试剂刺激细胞时,细胞中的Ras在前端被快速地激活(最高值在3~4 s 出现)[2],当Ras功能被阻止时,细胞不能发生极化,也不能感知环境中的化学信号发生定向运动,只发生随机运动。此外,PⅠ3Ks的趋化性功能依赖于趋化因子的浓度和盘基网柄菌细胞的发育阶段,可以说PⅠ3Ks作为一个复杂的细胞“罗盘”的组成部分,通过综合功能控制细胞的方向反应[3]。然而,研究者在利用PⅠ3K 突变株作为研究对象探讨趋化性机制的过程中,也曾观察到一些相矛盾的现象。例如:Buczynski 等[4]研究表明,PⅠ3K1 和PⅠ3K2 敲除的细胞株具有更快的趋化性,他们猜测,这个表型可能是由于pi3kA/B-细胞的骨架发生改变引起,也可能是影响了调节细胞趋化运动的Rho/Rac家族成员造成的;而Zhou 等[5]和Funamoto 等[6]则看到相反的现象,他们发现pi3kA/B-细胞的趋化性降低,速度也减弱。Loovers 等[7]研究发现,pi3kA/B-突变株发育5 h 表现出极化降低并且产生侧生伪足,但在其他发育条件下,该突变株具有与野生型细胞相同的行为。其他研究者利用LY294002 对不同类型细胞的研究也获得相互矛盾的结果[8],其中,用LY294002 短暂地处理盘基网柄菌细胞时,细胞不能有效地响应趋化因子,也不发生定向迁移,然而处理5 min甚至更长时间后,细胞发生极化并能够向高浓度趋化因子迁移[6-7],这些都暗示了PⅠ3Ks作用的复杂性。
细胞的趋电性也是近年来人们比较关注的细胞定向运动之一,它指的是细胞在直流电场中发生朝向电场正极或负极定向迁移的现象。许多类型的细胞都能响应直流电场,在电场中发生朝向阳极或者阴极的定向运动,例如:成纤维细胞[9]、恶性大鼠前列腺癌细胞[10]、沃克癌肉瘤细胞[11]、骨髓干细胞[12]、盘基网柄菌单细胞[13]等在直流电场中朝向阴极运动;人类粒细胞[14]、兔的角膜上皮细胞[15]、转移程度低的大鼠前列腺癌细胞AT-2[10]、人的血管内皮细胞[16]等朝向阳极运动;有些细胞随环境条件的变化,在电场中运动的方向会随着改变,甚至肿瘤细胞还会随恶性程度的增加,对电场的响应也增强,甚至迁移方向完全相反。近年来,在探究细胞趋电性运动机制过程中,研究者们关注到PⅠ3K对细胞趋电性运动的一些作用。Zhao等[17]研究发现,中性粒细胞缺乏PⅠ3Kγ的催化亚基p110的编码基因后,细胞的趋电性较野生型细胞显著降低;Meng 等[18]用PⅠ3K 的抑制剂处理胚胎和成年神经前体细胞,发现经PⅠ3K 抑制剂处理后两种细胞出现不同程度的趋电性改变。Sato 等[19]研究发现,在盘基网柄菌中,可溶性的鸟苷酸环化酶(sGC)和GbpC的催化结构域通过cGMP调节细胞的朝向阴极的信号,而sGC的N端结构域连同抑制PⅠ3K 以及cGMP 的产物调节着细胞朝向阳极的信号。但由于PⅠ3Ks种类和功能的复杂性,人们并不完全明确PⅠ3Ks与细胞趋电性中的作用机制。
在盘基网柄菌中,PⅠ3K1 和PⅠ3K2 都具有Ras结合结构域,它们都是细胞接收化学信号刺激后活化Ras 所必需的两个关键PⅠ3Ks[1]。为了揭示PⅠ3Ks在细胞趋电性中的作用,本研究对盘基网柄菌细胞中的PⅠ3K1和PⅠ3K2在细胞响应直流电场发生定向迁移中的作用进行研究。首先通过CRⅠSPR/Cas9 系统介导分别构建PⅠ3K1 的编码基因pikA和PⅠ3K2的编码基因pikB的单基因敲除突变株,其次对突变株的趋电性及其机制进行探讨,研究结果表明,PⅠ3K1 和PⅠ3K2 在盘基网柄菌细胞趋电性中发挥抑制作用,并且可能通过增强Akt和ERK的活性来发挥作用。
1 材料与方法
1.1 材料
细胞:野生型盘基网柄菌细胞AX2;pikA-突变株;pikB-突变株;克雷伯氏菌(Klebsiella aerogenes,KA);大肠杆菌感受态细胞(DH5α)。
质粒:pTM1285(CRⅠSPR/Cas9 基因编辑载体,来自日本NBRP Nenkin(National BioResource Project Cellular slime molds)。
分子克隆酶制剂:BbsⅠ-HF (R3539L,BioLabs公司);rTaq酶;T4 DNA连接酶(M0202S,BioLabs公司)。
抗体:P44/42 MAPK(ERK1/2) Rabbit mAb(HRP Conjugate)(CST);Phospho-p44/42 MAPK(ERK1/2) (Thr202/Tyr204) (D13.14.4E) XP®Rabbit mAb(CST);GADPH 抗体(Rabbit pAb,30202ES40,翌圣生物科技(上海)股份有限公司);T-Akt (pan)(C67E7) Rabbit mAb #4691(CST) ; Phospho-Akt Substrate (RXXS*/T*)(110B7E) Rabbit mAb #9614 (CST) ;Anti-β-tubulin Antibody(BOSTER,A01857-1)。
分子试剂盒:TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver. 4.0 (TaKaRa 公司);TaKaRa MiniBEST Universal Genomic DNA Extraction Kit Ver.5.0 (TaKaRa 公司);TaKaRa MiniBEST Plasmid Purification Kit Ver.4.0(TaKaRa公司)。
引物合成及测序:引物合成和测序均由北京擎科新业生物技术有限公司完成。
HL5培养基:葡萄糖10 g,示蛋白胨10 g,酵母粉5 g,磷酸氢二钠0.96 g,磷酸二氢钾0.48 g,硫酸链霉素0.03 g,蒸馏水定容到至1 L,pH 6.5,121℃湿热灭菌30 min。
H50 缓冲液:20 mmol/L HEPES(4-羟乙基哌嗪乙磺酸),pH 7.0;50 mmol/L氯化钾,10 mmol/L氯化钠,1mmol/L 硫酸镁,5 mmol/L 碳酸氢钠,1 mmol/L磷酸二氢钠,过滤除菌。
发育缓冲液(DB):先配制10×磷酸盐缓冲液(磷酸氢二钠12.695 g,磷酸二氢钾6.803 g,水定容至1L)。使用时配制用蒸馏水稀释至1×DB(100 ml 10×磷酸盐缓冲液、1 mol/L 氯化钙0.2 ml、2 mol/L硫酸镁1 ml,水定容至1 L)。
Steinberg’s 溶液:氯化钠 580 mmol/L,氯化钾6.7 mmol/L, 硝酸钙4.4 mmol/L, 硫酸镁13 mmol/L,三羟甲基氨基甲烷46 mmol/L,水定容至1 L,pH 7.5~7.7,使用时用水稀释10倍。
LB 培养基:示蛋白胨10 g/L,酵母粉5 g/L,氯化钠10 g/L;必要时加入琼脂粉15 g/L(固体)和/或终浓度为100 mg/L的氨苄青霉素钠。
SM 培养基:示蛋白胨10 g/L,酵母粉1 g/L,葡萄糖10 g/L,硫酸镁1 g/L,磷酸氢二钾1.9 g/L,磷酸二氢钾0.6 g/L,琼脂粉20 g/L (固体),pH 6.4,121℃灭菌30 min。
1.2 方法
1.2.1sgRNA设计、二聚化和磷酸化
从dictybase.org 上获取盘基网柄菌pikA和pikB的基因序列(pikA: DDB_G0278727;pikB:DDB_G0274109),利用网站http://crispr.dbcls.jp/分别设计pikA和pikB的sgRNA(single-guide RNA),相关引物信息见表1。
用灭菌蒸馏水将合成的sgRNA 正向与反向引物稀释至100 μmol/L,分别取1 μl至PCR管,用灭菌蒸馏水将反应体系体积补充至10 μl,于PCR 仪中用95℃、5 min;Ramp -2.5%、25℃程序使引物二聚化。随后,取5 μl 引物二聚体到灭菌离心管中,并加入1 μl T4 PNK 和1 μl T4 PNK Buffer,并用灭菌双蒸水将反应体系补充至10 μl,于37℃水浴中严格处理30 min,连接或放入-80℃冰箱保存。

Table 1 Primer information
1.2.2pTM1285-sgRNA载体的制备
用BbsⅠ内切酶消化环状质粒pTM1285 使之形成线性的质粒骨架后,酶切体系中加入等体积的异丙醇,混匀并转移到质粒提取收集柱中,用含75%乙醇的PE 溶液纯化和双蒸水洗脱,获得纯化的线性质粒。构建pTM1285-sgRNA 载体时,将2 μl 稀释100 倍的磷酸化引物二聚体、2 μl 线性的pTM1285 质粒、1 μl 10×T4 Ligase Buffer、1 μl T4 Ligase混合,并用灭菌双蒸水将反应体系体积补充至10 μl,于16℃水浴中连接过夜。42℃热激法将连接产物转化至大肠杆菌DH5α感受态细胞中,涂板到LB 固体培养基(Amp+)筛选16~20 h。挑取若干克隆子,利用反向的sgRNA引物和tRNA进行菌落PCR 鉴定(表1)。经测序验证后提取相应克隆子中的质粒用于电转化实验。
1.2.3电转化pTM1285-sgRNA载体
无菌培养野生型盘基网柄菌AX2 细胞至指数生长期(方法见参考文献[20]),收集2×107个AX2 细胞至15 ml 离心管中,冰上处理15 min,其间颠倒翻转多次,于4℃、2 000 r/min 条件下离心5 min,弃上清液;再用预冷的H50缓冲液离心洗2次;最后用100 μl H50 重悬细胞,并加入5 μl pTM1285-sgRNA 载体与之混合后,将体系转入0.1 cm的电击杯中,用电穿孔仪进行电击转化,条件为750 V、25 μF,电击2 次,2 次电击之间间隔5 s,以不加载体的体系为对照。电击结束后,将悬液全部转入10 ml HL5 培养基培养24 h。随后向培养基中加G418(20 g/L)进行筛选,约3 d 时,对照组细胞开始凋亡变圆,而实验组中的克隆子开始贴壁生长。
1.2.4噬菌斑分离单克隆
用SM 平板活化KA后,挑取单菌落到100 ml液体SM培养基中,22℃培养48 h可达到实验需要的菌体密度,该菌液可以使用7 d左右。
离心收集电转化后经过G418 筛选3 d 的克隆子,于1.5 ml离心管中加入1 mlKA菌液使之重悬,依次吸取10、20、50、100、200、500 μl的悬液分别和990、980、950、900、800、500 μl的KA菌液混合后,涂布到SM固体平板上,于22℃培养箱中倒置培养,一般3~5 d 开始形成噬菌斑,每1 个独立的噬菌斑即为1个单克隆。
噬菌斑形成后,分别挑取噬菌斑周围的细胞到24 孔板中培养,每孔加入2 ml HL5 培养基(含双抗和G418),22℃培养3 d 后,阳性克隆子开始迅速生长,随后这些细胞将被转移到细胞培养皿中扩大培养,此时,从每1个噬菌斑中分离获得的细胞被定义为1个突变株。
1.2.5突变株的DNA测序鉴定和蛋白质印迹鉴定
当突变株细胞扩增到足够量后,用基因组DNA 提取试剂盒分离突变株的基因组DNA,并以此DNA为模板,用引物screen-F和screen-R(表1)扩增获得单一片段。切胶回收、纯化目的片段后进行测序,获得的序列与野生型细胞中的序列比对,由此可知突变信息。
用蛋白质印迹法检测突变株细胞中的AKT 和ERKA蛋白的表达情况及其活性(方法详见参考文献[21])。
1.2.6细胞的趋电性研究方法
细胞趋电性运动的研究方法参考文献[21]。简述如下:
无菌培养AX2 细胞和突变型细胞至指数生长期,取适量细胞于24 孔板中,22℃使细胞贴壁;用新鲜的DB缓冲液轻轻洗2次后,继续用DB缓冲液饥饿处理3 h。重悬细胞,取适量细胞种到加电小室中,贴壁5 min,盖上盖玻片,加入适量的DB缓冲液,根据实验目的,在小室两端用导电盐桥连接直流电场。用带有CCD 的显微镜记录细胞在电场中的运动情况,拍照频率为间隔1 min 拍1 帧,共30 min。拍照结束后,将图片导入ⅠmageJ 软件中进行分析。每组实验至少重复3次,每次分析的细胞个数为50 个。统计结果用方向性指数(directedness)和方向持续性指数(persistency)评价细胞在直流电场中的方向及在某一特定方向上的持续行为;用细胞的轨迹速度(track speed)和位移速度(displacement speed)评价细胞在直流电场中的运动能力,其中,轨迹速度指的是细胞在单位时间(min)内的运动路程(μm),位移速度指的是单位时间(min)内的位移路程(μm)。
2 结果
2.1 CRISPR/Cas9系统构建基因编辑载体
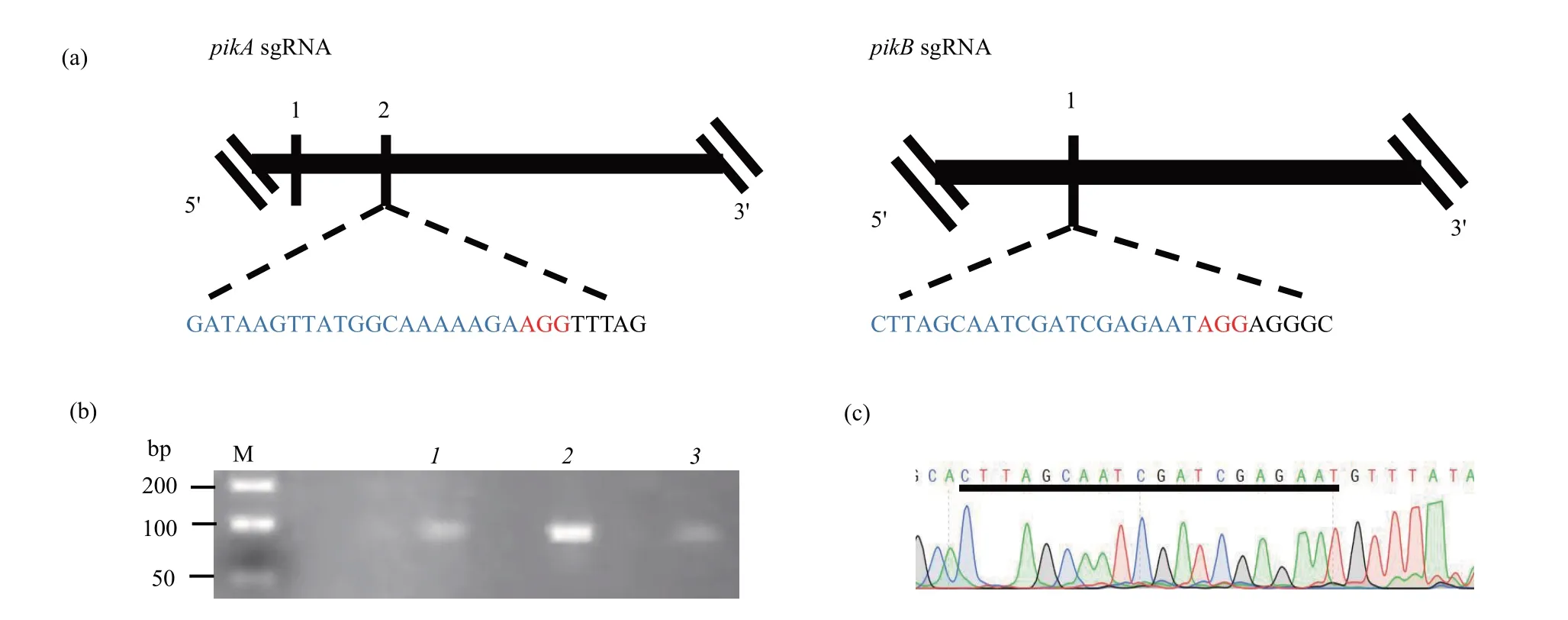
Fig. 1 Selection of sgRNA and plasmid construction
本研究选用all-in-one 表达载体pTM1285 作为质粒骨架构建CRⅠSPR/Cas9基因编辑载体。先根据pikA和pikB基因的序列信息,在外显子序列中寻找PAM 序列(NGG),选取该序列上游紧邻的20 bp碱基序列作为sgRNA(图1a),并通过在NCBⅠ系统中BLAST 比对,排除单核苷酸多态性序列的可能性,并最终确定sgRNA 的序列信息(表1)。根据sgRNA 在pTM1285 质粒中的插入位点,在sgRNA 的末端加上BbsⅠ酶切位点序列AGCA(正义链5'端)和AAAC(反义链5'端)。将引物二聚化和磷酸化后,与BbsⅠ酶切消化的线性pTM1285质粒骨架连接,热激转化到大肠杆菌DH5α中,并用氨苄青霉素钠抗性筛选。以转化子为模板,利用sgRNA的反向序列和tRNA的正向序列为引物进行PCR 扩增,可获得约100 bp 的扩增片段(图1b),该扩增产物经纯化后对其进行测序(图1c),根据序列信息即可鉴定质粒是否已经重组。提取经测序鉴定的转化子中的质粒DNA,获得pTM1285-sgRNA-pikA和pTM1285-sgRNA-pikB重组载体。
2.2 突变株的分离
通过电穿孔技术将pTM1285-sgRNA-pikA和pTM1285-sgRNA-pikB重组载体分别导入野生型AX2细胞中,利用20 mg/L G418筛选3 d后,阳性转化子开始分裂增殖,此时,收集转化子并与KA混合后逐级稀释涂布到SM 平板上,约3 d 后,噬菌斑开始出现(图2a),随着培养时间的延长,噬菌斑也进一步变大,而当噬菌斑中央区域的绝大部分KA被盘基网柄菌细胞摄取后,细胞开始处于饥饿状态,并开始转向多细胞发育。在突变株细胞筛选过程中,也可以初步根据噬菌斑中多细胞发育的表型进行初筛。例如,当靶基因是参与盘基网柄菌多细胞发育的关键基因时,噬菌斑中在多细胞发育情况也会随之发生变化,出现一些发育缺陷,例如聚集缺失、孢子囊较小、噬菌斑边缘不整齐等。进行扩大培养。随后,通过提取这些细胞的基因组DNA,并利用靶向序列两端的引物进行扩增(图2b)。扩增片段的大小可能与野生型中靶向序列接近,也有可能差异较大,因此,只要获得单一特异性的扩增片段,都将进一步对其开展测序鉴定。在本项研究中,首先基于蛋白质三联密码子的移码突变,本研究选择碱基序列变化为非三整数倍的突变类型,其次再通过蛋白质结构预测,最终在本研究中分别选择了pikA--1(代表pikA基因的突变) 和pikB--5(代表pikB基因的突变)两个突变株作为研究细胞趋电性的突变株。其中,与野生型的pikA基因序列相比,pikA-1的基因序列缺少了一个碱基,而与野生型的pikB基因序列相比,pikB--5 的基因序列则增加了4个碱基(图2c)。
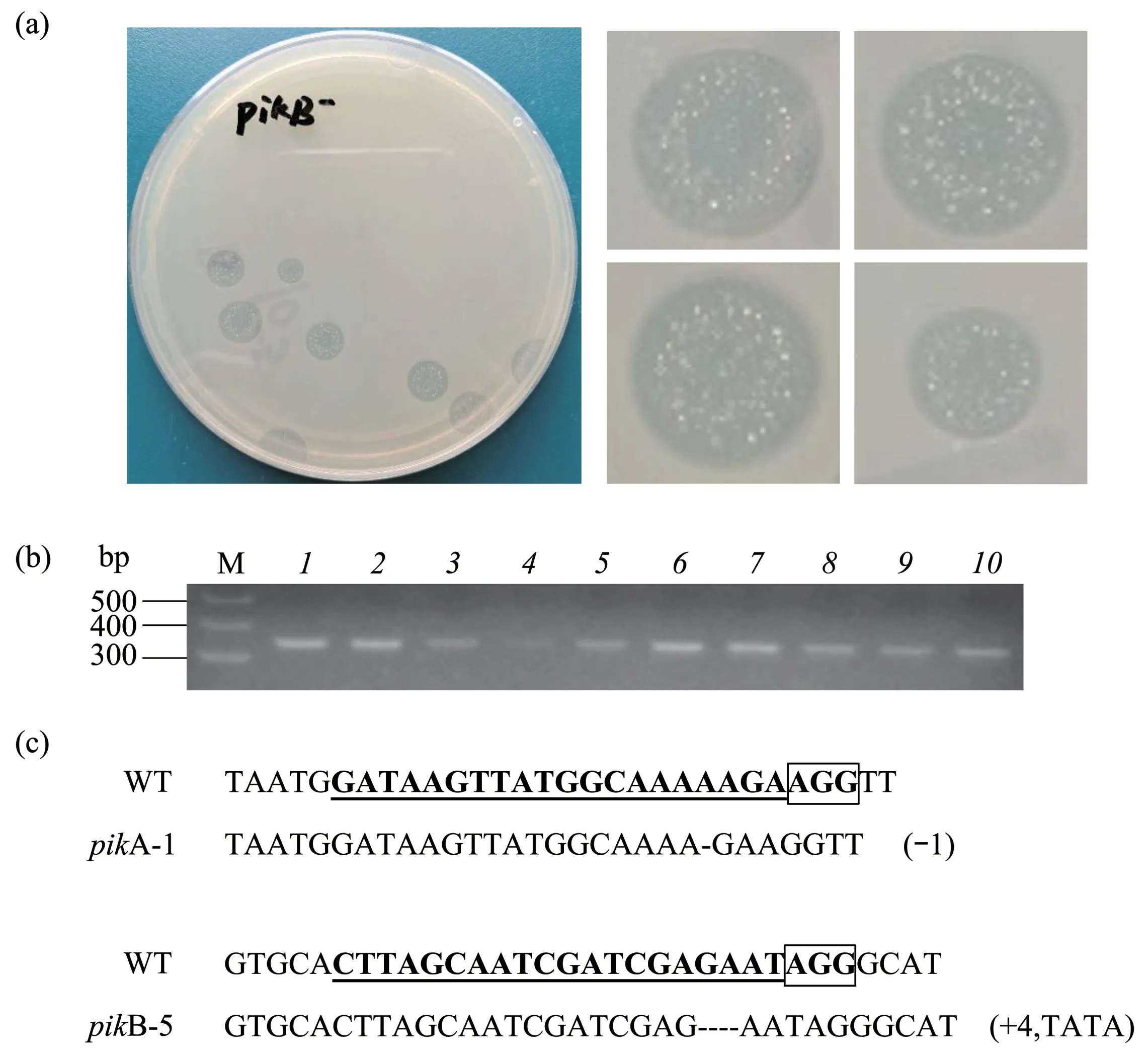
Fig. 2 Identification of gene knockout cell lines
2.3 多细胞发育鉴定
野生型盘基网柄菌细胞在饥饿条件下会形成具有孢子囊结构的多细胞有机体,这种从单细胞形成多细胞的过程被称为多细胞发育。为了研究方便,研究者认为的将盘基网柄菌的多细胞发育分为细胞聚集阶段(约4 h)、细胞丘阶段(约8 h)、蛞蝓体阶段(约12 h)和子实体阶段(约22 h)。
为了探究pikA和pikB基因在盘基网柄菌多细胞发育过程中的作用,本研究将pikA和pikB基因缺失突变株置于无营养的琼脂培养基上,分别涂布6.5×105/cm2的野生型细胞、pikA突变株细胞和pikB突变株细胞。22℃条件下倒置培养,并利用体视显微镜分别观察培养8 h 和24 h 时三种细胞的多细胞发育情况(图3)。从实验所获得的照片来看,饥饿8 h的野生型细胞AX2和pikA突变株都已经处于细胞丘阶段,但pikA突变株还有少数细胞处于聚集阶段,与二者相比,pikB突变株细胞的发育要延迟一些,此时的pikB突变株细胞发育才进入聚集阶段。当饥饿24 h后,野生型细胞和突变株细胞都形成了具有孢子囊结构的多细胞有机体,然而,与野生型细胞形成的多细胞有机体相比,pikA突变株细胞和pikB突变株细胞形成的多细胞体在形态和大小上都表现出差异性。其中,pikA-突变株细胞形成的孢子囊较野生型细胞形成的孢子囊结构偏长,孢子梗更粗壮,能将孢子囊直立于空气中,而pikB-突变株细胞形成的孢子囊形状与野生型相似,但却比野生型细胞形成的孢子囊更小,孢子梗较纤细,使得大部分的孢子囊倒在基质表面。这些结果暗示了PⅠ3K1和PⅠ3K2在盘基网柄菌多细胞发育过程中发挥的功能是有差异的。
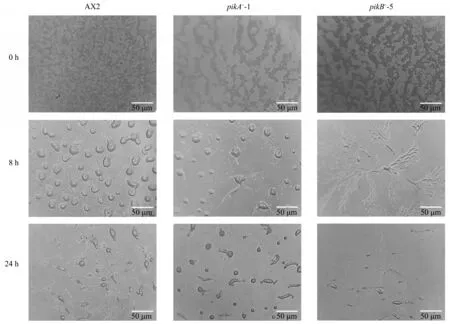
Fig. 3 Development of wild-type and mutant cells plated on non-nutrient agar
2.4 突变株在直流电场中的趋电性
将野生型细胞和突变株细胞分别置于场强为12 V/cm的直流电场中,通过实时录像系统记录细胞的运动轨迹,结果发现,pikA-和pikB-突变株在直流电场中的运动方向与野生型细胞相同,都是朝向电场负极方向运动(图4a,b)。用软件对细胞在直流电场的趋电性进行分析,结果发现:野生型细胞在直流电场中的运动方向指数为(0.86±0.03),而pikA-和pikB-突变株在直流电场中的运动方向指数分别为(0.95±0.02)和(0.94±0.03),二者与野生型细胞之间存在显著差异;野生型细胞的方向持续性指数为(0.36±0.01),pikA-和pikB-突变株的方向持续性指数分别为(0.60±0.01) 和(0.62±0.01),说明与野生型细胞相比,pikA-和pikB-突变株在直流电场朝向负极运动轨迹更为“笔直”。对三种细胞在直流电场中的运动能力进行统计分析发现,野生型细胞的平均轨迹速度(3.34±0.08)μm/min,而pikA-和pikB-突变株的平均轨迹速度分别为(4.85±0.20)和(5.48±0.15)μm/min,t检验表明突变株和野生型之间存在极显著的差异。平均位移速度统计分析结果显示,野生型细胞的平均位移速度(1.26±0.05)μm/min,而pikA-和pikB-突变株的平均位移速度分别为(3.04±0.17)和(3.46±0.13)μm/min,t检验同样表明突变株和野生型之间存在极显著的差异(图4c)。以上结果揭示了pikA和pikB基因在盘基网柄菌细胞响应直流电场发生定向迁移过程中发挥着抑制作用,当人为干扰pikA和pikB基因后,突变株对相同强度的直流电场中的响应更为强烈,此外,pikA和pikB基因还抑制了电场作用下的细胞运动能力。
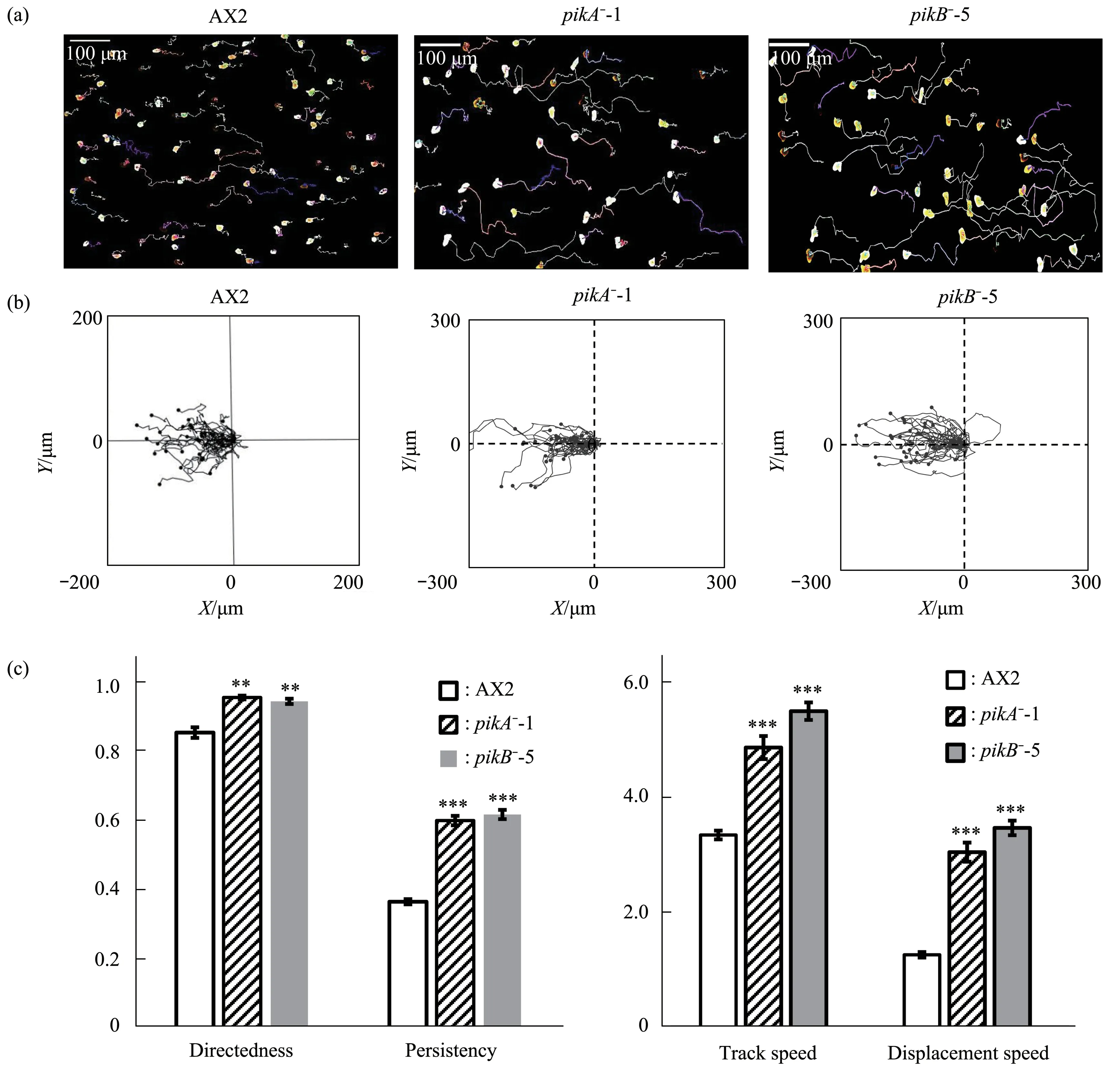
Fig. 4 Electrotaxis of pikA and pikB mutants
2.5 PI3K1和PI3K2通过调节Akt和ERK抑制细胞的趋电性
据报道,盘基网柄菌细胞的运动速度和趋电性分别与Akt 和ERKB 有关,因此,为了初步探究pikA和pikB基因缺失突变株在直流电场中的响应机制,本研究利用蛋白质印迹技术分别探究了pikA和pikB突变株中的Akt 和ERK 的蛋白质表达情况。结果显示(图5),与野生型AX2中p-Akt的表达量相比,pikA和pikB突变株中的p-Akt的表达显著增加,尤其是pikB突变株中的p-Akt的表达更高,表明细胞中高表达的p-Akt可能是细胞运动速度增加的一个关键原因。此外,两个突变株中ERK 的表达也与野生型明显不同,pikA和pikB突变株中的p-ERKA 降低,而p-ERKB 的表达量却增加了,推测细胞中p-ERKB的高表达可能是抑制细胞响应电信号的原因之一。
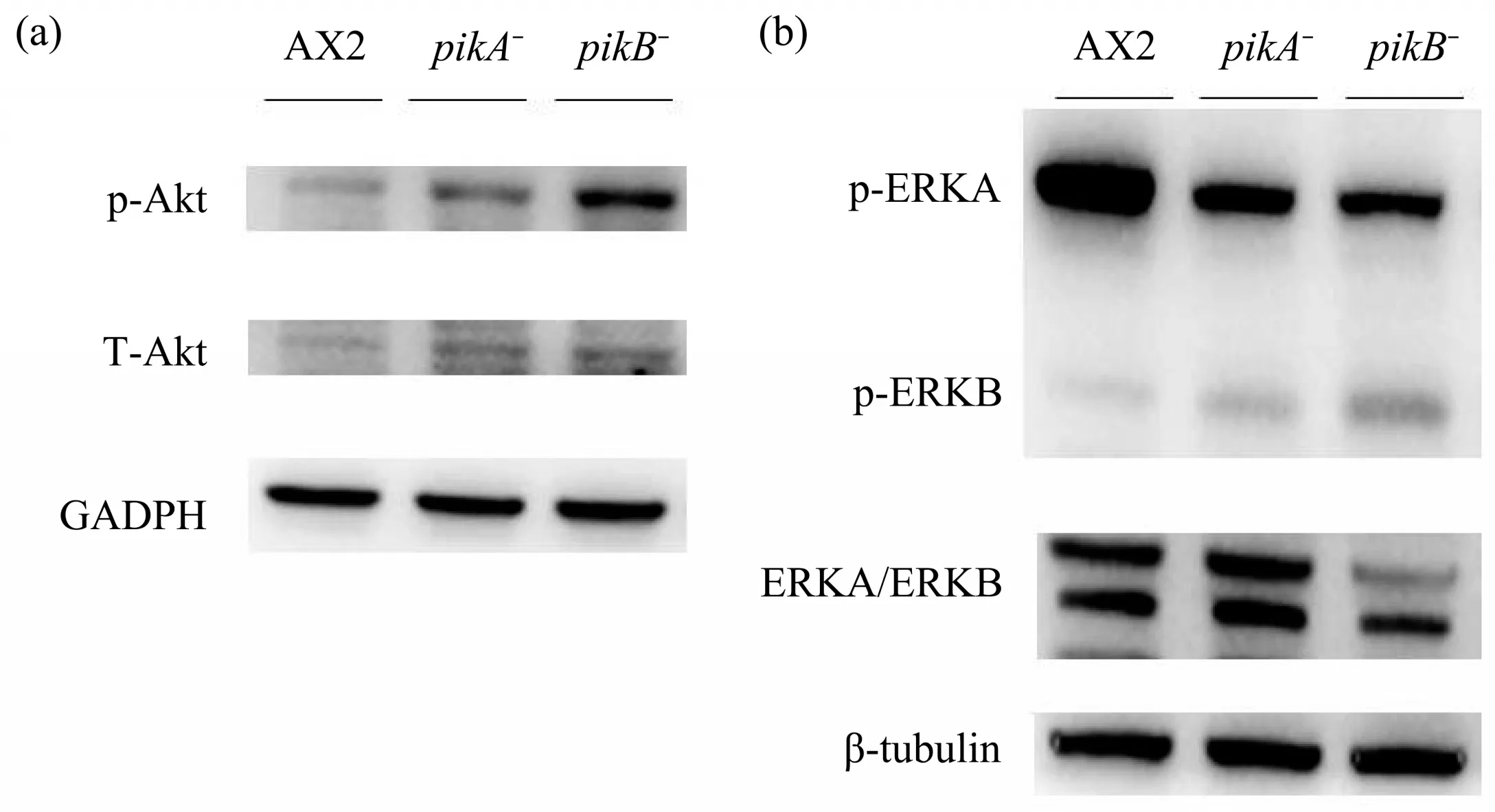
Fig. 5 Western blot showed expression of Akt and ERK in wild-type and mutant cells
3 讨论
PⅠ3K 是细胞定向迁移中一个关键的信号分子[17,22]。基因组全序列测序分析发现,盘基网柄菌中共有6 个Ⅰ类的PⅠ3K,其中PⅠ3K1~5(编码基因依次为pikA、pikB、pikC、pikF、pikG)具有较长的N 端结构域,1 个Ras 结合结构域(RBD)、1个C2结构域、1个激酶辅助结构域,以及1个激酶催化结构域。从激酶结构域组成分析,盘基网柄菌中的PⅠ3K1 和PⅠ3K2 分别与人类的Ⅰa 和Ⅰb PⅠ3Ks 最为相似[3],功能上有相同部分但也存在差异,例如,Buczynski 等[4]研究发现,盘基网柄菌的PⅠ3K1 和PⅠ3K2 在调节盘基网柄菌的内吞、细胞形状和细胞运动中发挥不同但有选择性的作用。近来,Kim 等[23]研究表明,同时缺失PⅠ3K1 和PⅠ3K2的盘基网柄菌细胞,其运动速度较野生型细胞显著增加,暗示了这个两个蛋白质在细胞运动中的抑制作用,然而,并未明确哪个蛋白质在细胞速度的调控方面发挥着更为主导的作用。本项研究通过单基因敲除研究发现,PⅠ3K1 和PⅠ3K2 在盘基网柄菌细胞趋电性运动中可以独自发挥抑制作用,其中包括运动的速度和对电场的响应两方面,本研究中发现的突变株多细胞发育差异也可以为该推测提供一定的实验证据,该结果暗示了两个蛋白质在细胞信号网络中发挥着不同的功能,可能是与不同的Ras蛋白作用激活不同的信号通路。
成簇的规律间隔短回文重复序列(CRⅠSPR)相关蛋白9核酸酶(Cas9)介导的基因组编辑是一个强大的基因组编辑工具,该技术已被广泛用于功能基因组研究。在通常使用的CRⅠSPR/Cas9 系统中,sgRNA的表达由RNA聚合酶ⅠⅠⅠ依赖的启动子(如U6)产生。盘基网柄菌属U6核内小RNA被认为是由RNA聚合酶ⅠⅠⅠ转录的,因为它缺乏在其他生物体中观察到的U6 的三甲基化5'帽。此外,U6启动子的使用需要一个“G”残基作为sgRNA的第一个碱基来启动转录,这限制了合适靶序列的数量。因此,利用内源tRNA加工系统在盘基网柄菌属中表达sgRNA。tRNA序列之间插入sgRNA会引起RNase P 和RNase Z 识别三叶草结构并在tRNA的5'和3'端切割,产生成熟的sgRNAs。本文基于CRⅠSPR/Cas9基因编辑技术,利用适用于盘基网柄菌的all-in-one 表达载体pTM1285,构建了pikA和pikB的基因突变株,为利用CRⅠAPR/Cas9 基因编辑技术构建盘基网柄菌细胞的其他基因突变株提供了参考。
4 结论
本研究表明,盘基网柄菌细胞中的PⅠ3K1 和PⅠ3K2在细胞响应直流电场发生定向迁移中各自发挥着抑制作用,具体体现在趋电性指数的显著增加,同时在细胞运动速度上,PⅠ3K1和PⅠ3K2也各自发挥着抑制作用,这种抑制作用可能与细胞中的Akt 和ERKB 的高表达相关,但具体的作用机制还需进一步研究。
