超高效液相色谱-三重四级杆串联质谱法测定化妆品中达克罗宁和新康唑
2024-03-19吴旭金鹏刘洋邢海艳吴义春孙俪苏晶
吴旭,金鹏,刘洋,邢海艳,吴义春,孙俪,苏晶
(淮安市食品药品检验所,江苏淮安 223300)
新康唑是一种咪唑类新型广谱抗生素[1],具有去屑、杀菌和调节皮肤油脂[2]的作用,在我国暂未列入《已使用化妆品原料名称目录》,在欧洲化妆品领域中作为去屑剂使用[3]。达克罗宁是一种处方类局部麻醉药,起效快、持续久,通过阻断神经冲动或刺激传导,抑制触觉和痛觉,具有止痛、止痒及杀菌的作用[4]。美国药典第35 版已有收录,我国药典2020版还未收录,在我国临床已广泛使用多年,有胶浆剂[5]、乳膏剂[6]、洗剂[7]等种剂型,多用于黏膜表面麻醉[8]及皮肤镇痛、止痒[9]。一些不法商家在化妆品中非法添加新康唑和达克罗宁,使化妆品具有快速去湿疹、去红血丝、止痒等使用效果。长期使用添加新康唑和达克罗宁的化妆品,会对人体健康产生影响[10-12],特别是药物滥用会给儿童带来的巨大危害[13-14]。
新康唑和达克罗宁在化妆品中的检测研究较少,已知检测方法包括紫外分光光度法[15]、高效液相色谱法[16]等。为了更好地服务化妆品安全监管,规范化妆品中禁限用物质检测技术要求,确保化妆品安全质量,笔者采用超高效液相色谱-三重四级杆串联质谱技术,建立化妆品中新康唑和达克罗宁非法添加物定性、定量检测方法,提高化妆品非法添加物检测效率、减少基质干扰、节约检测成本,并进行方法的验证及应用研究。
1 实验部分
1.1 主要仪器与试剂
三重四极杆质谱仪:AB SCⅠEX APⅠ 4000+型,配有V型Turbo电离源,美国AB SCⅠEX公司。
超高效液相色谱仪:Waters Acquity UPLC HClass型,美国沃特世公司。
电子天平:XS205D 型,感量为0.01 mg,瑞士梅特勒-托利多科技有限公司。
高速低温离心机:Thermo MULTⅠFUGE X1R型,美国赛默飞世尔科技公司。
超声波清洗仪:KH7200 型,昆山禾创超声仪器有限公司。
涡旋混合仪:XW-80A 型,上海驰唐电子有限公司。
超纯水机:Milli-QR型,美国密理博公司。
甲醇、乙腈、甲酸:均为色谱纯,美国默克公司。
氯化钠:分析纯,国药集团化学试剂有限公司。
饱和氯化钠溶液:称取40 g 氯化钠,置于250 mL烧杯中,加入100 mL水,超声15 min。
盐酸达克罗宁:质量分数(按C18H27NO2·HCl计)为99.8%,批号为100423-201102,于105 ℃干燥3 h后使用,中国食品药品检定研究院。
新康唑:质量分数为99.8%,批号为1028-RC-0061,上海源叶生物科技有限公司。
化妆品样品:爽肤水、凝胶、润肤霜、精华油、润肤乳5种化妆品,市售。
1.2 仪器工作条件
1.2.1 色谱条件
色 谱 柱:Waters ACQUⅠTY UPLC®BEH C18柱(100 mm×2.1 mm,1.9 μm,美国沃特世公司);柱温:30 ℃;进样体积:2 μL;流动相:A相为0.1%(体积分数)甲酸溶液,B 相为甲醇,梯度洗脱程序见表1,流量为0.3 mL/min。
1.2.2 质谱条件
离子源:电喷雾离子源(ESⅠ源);监测模式:正离子多反应监测模式(MRM);监测离子对及相关参数设定见表2。
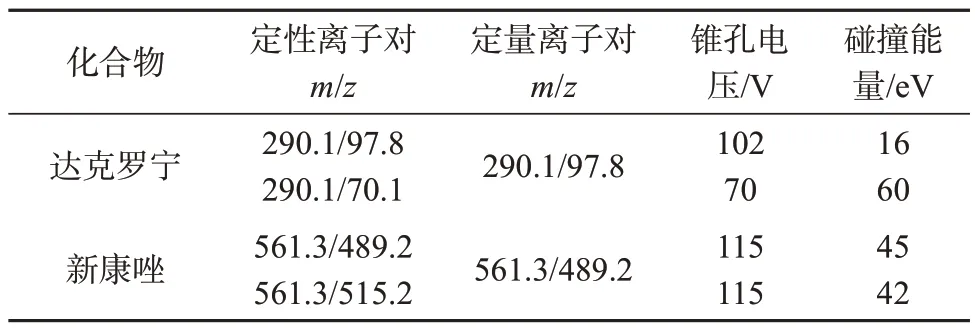
表2 达克罗宁和新康唑质谱分析参数Tab.2 Mass analysis parameters of dyclonine and elubiol
1.3 溶液制备
1.3.1 标准储备溶液
精密称取达克罗宁、新康唑标准品各10 mg(精确到0.01 mg),分别置于10 mL棕色容量瓶中,加入乙腈使溶解并定容至标线,摇匀。所得达克罗宁、新康唑标准储备液的质量浓度均为1 000 mg/L。
1.3.2 混合标准储备溶液
准确移取达克罗宁、新康唑标准储备溶液各1.0 mL,置于100 mL 棕色容量瓶中,用乙腈稀释定容至标线,摇匀,于-18 ℃冰箱中避光保存。该混合标准储备溶液中两组分的质量浓度均为10 mg/L。
1.3.3 样品溶液
称取化妆品样品0.2 g(精确至0.1 mg),置于20 mL具塞比色管中,加入饱和氯化钠溶液2 mL,涡旋振荡30 s,加入乙腈1 mL,涡旋30 s,再加入乙腈10 mL,涡旋30 s,置冰浴中超声提取30 min,以4 000 r/min 转速离心10 min。将乙腈层移入另一20 mL具塞比色管中,残渣再加入乙腈8 mL,重复提取一次,合并两次乙腈提取液,并用乙腈定容至标线,取上述提取液用0.22 μm 有机微孔滤膜过滤,滤液作为样品溶液。
1.3.4 空白基质提取溶液
称取空白化妆品样品0.2 g(精确至0.1 mg),置于20 mL具塞比色管中,与样品溶液制备同法处理,作为空白基质提取液。
1.3.5 基质混合标准中间液
准确量取混合标准储备溶液1.0 mL,置于10 mL棕色容量瓶中,用空白基质提取液稀释至标线,摇匀,制成达克罗宁、新康唑质量浓度均为1 000 μg/L的基质混合标准中间液,应现用现配。
1.3.6 系列基质混合标准工作溶液
移取基质混合标准中间液适量,用空白基质提取溶液稀释,制得达克罗宁、新康唑质量浓度均分别为1、5、10、20、50、100 μg/L 的系列基质混合标准工作溶液,应现用现配。
1.4 实验步骤
在1.2仪器工作条件下,将系列基质混合标准工作溶液分别进样测定,以目标物的质量浓度为横坐标,对应色谱峰面积为纵坐标,进行线性回归,建立基质标准曲线。取样品溶液进样测定,将对应目标物色谱峰面积代入基质标准曲线,得到目标物的质量浓度,再计算得到样品中目标物的质量分数。
2 结果与讨论
2.1 样品处理方法讨论
2.1.1 提取溶剂的选择
化妆品基质复杂,达克罗宁和新康唑在甲醇、乙腈中的溶解作用良好。以空白加标样品为对象,比较了饱和氯化钠-乙腈、乙腈和甲醇三种提取溶液对两种目标物的提取效率,结果见表3。
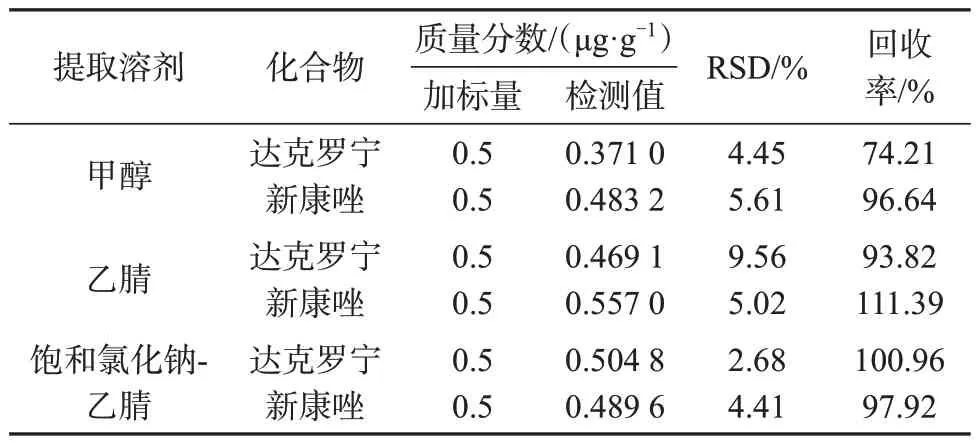
表3 不同提取溶剂提取回收率(n=6)Tab.3 Recovery rates of different solvents extraction (n=6)
由表3数据可知,乙腈或饱和氯化钠-乙腈提取溶液对两种目标物的提取回收率均较高,但乙腈提取后的样品不易完全溶解,有结块,而添加饱和氯化钠溶液可避免样品起泡,有利于样品破乳溶解,对两种目标物的提取效率均良好,因此选择采用饱和氯化钠-乙腈作为提取溶剂。
2.1.2 提取方式和提取条件的选择
以阳性样品为考察对象,分别在冰浴、室温(25 ℃)、60 ℃三种提取温度下超声提取30 min,以及在冰浴下超声提取0、10、20、30、40 min,考察不同提取方式的目标物提取率,结果列于表4和表5。
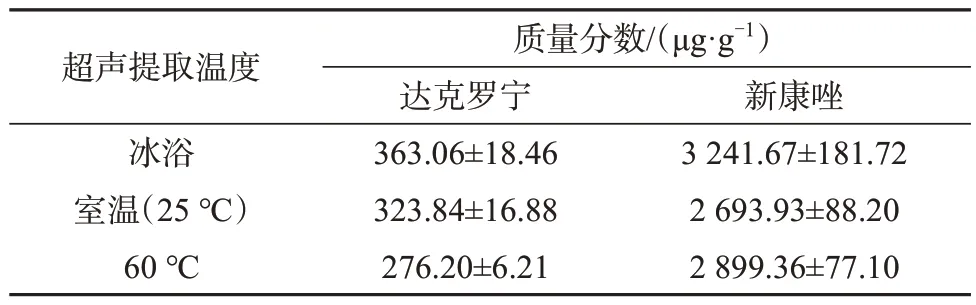
表4 不同超声提取温度下目标物测定值(n=6)Tab.4 Test results of different ultrasonic extraction temperatures
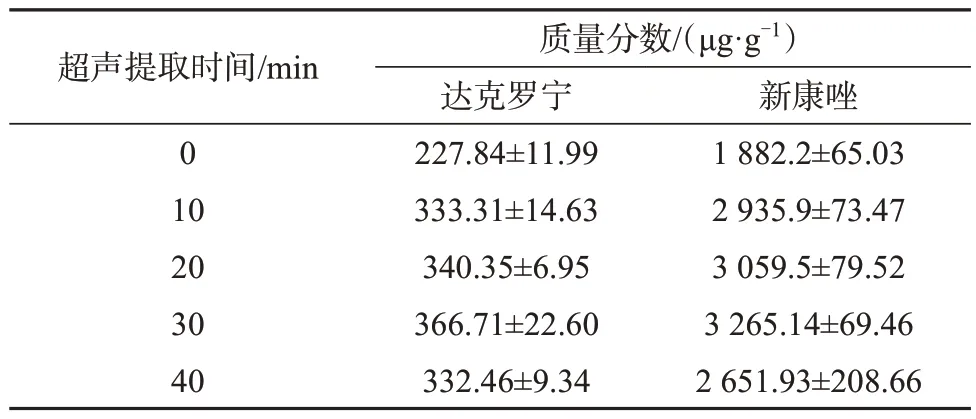
表5 不同超声提取时间的目标物测定值(n=6)Tab.5 Test results of different ultrasound extraction times
由表4可知,随着提取温度增加,目标物测定值减小,因此确定采用冰浴提取样品。由表5可知,冰浴条件下两种目标物测定值随提取时间的延长而增大,当提取时间超过30 min后有所减小。综合考虑提取效率和提取时间,选择超声提取时间为30 min。最终选择冰浴条件下超声30 min来提取目标物。
2.2 色谱条件
2.2.1 液相色谱柱的选择
实验比较了Waters ACQUⅠTY UPLC®BEH C18色谱柱(100 mm×2.1 mm,1.9 μm,美国沃特世公司)和Thermo Hepersil Gold C18色谱柱(100 mm×2.1 mm,1.7 μm,美国赛默飞世尔科技公司)2种不同品牌类型的C18色谱柱对目标化合物峰型及分离效果的影响,结果表明,二者均能够满足分析要求,由于Waters ACQUⅠTY UPLC®BEH C18色谱柱出峰时间更快,所以选择其作为实验用色谱柱。
2.2.2 流动相的选择
参考《化妆品安全技术规范》,分别采用甲醇-0.1%甲酸水溶液、甲醇-水和乙腈-0.1%甲酸水体系作流动相,比较了目标化合物的色谱峰形及分离效果,结果表明,在三种流动相体系下目标化合物均可得到有效分离,且色谱峰形良好。考虑到加入适量酸可以提供质谱的离子化效果,使用甲醇可节约成本,最终选择甲醇-0.1%甲酸水溶液作为流动相。
2.3 质谱条件
设定色谱流动相流量为0.3 mL/min,按照仪器厂家推荐设置气流、源温、毛细管电压等参数(喷雾压力:207 kPa;干燥气压力:379 kPa;干燥气温度:500 ℃;毛细管电压:5 500 V),利用三重四极杆质谱定量和定性分析,针对每个目标物分别进样进行质谱参数的优化,主要包括定性和定量离子对、锥孔电压和碰撞能量的选择。从一级质谱扫描图和二级碎裂质谱扫描图可知,达克罗宁在ESⅠ正离子模式下,形成[M+H]准分子离子峰290.1,主要碎片离子质荷比为97.8、70.1;新康唑准分子离子峰质荷比为561.3,主要碎片离子质荷比为515.2、489.2。采用仪器优化程序对上述离子对的锥孔电压和碰撞能量值进行优化,最终确定达克罗宁锥孔电压分别为102、70 V,碰撞能量分别为16、60 eV,新康唑锥孔电压分别为115、115 V,碰撞能量分别为45、42 eV。
2.4 专属性
经过处理的空白化妆品样品、空白化妆品样品加标对照品,在选定的色谱条件下注入液相色谱-质谱联用仪进行分析,色谱图见图1。
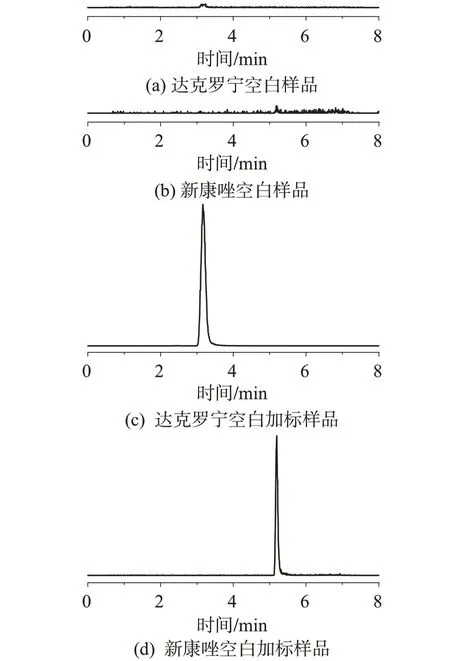
图1 空白样品和空白加标样品的UHPLC-MS/MS色谱图Fig.1 UHPLC-MS/MS chromatograms of blank sample and blank spiked sample
由图1可见,化妆品基质对2种待测物质测定无干扰,说明所建方法适用于化妆品中两种目标物的测定。
2.5 基质效应
分别选择爽肤水、凝胶、润肤霜、精华油、润肤乳5种典型的空白化妆品作为基质,考察基质效应,结果见表6。由表6数据可以看出,达克罗宁和新康唑在化妆品中存在基质效应,低含量时基质效应影响较小,而高含量时基质效应显著,所以采用基质标准曲线作为定量校准曲线。
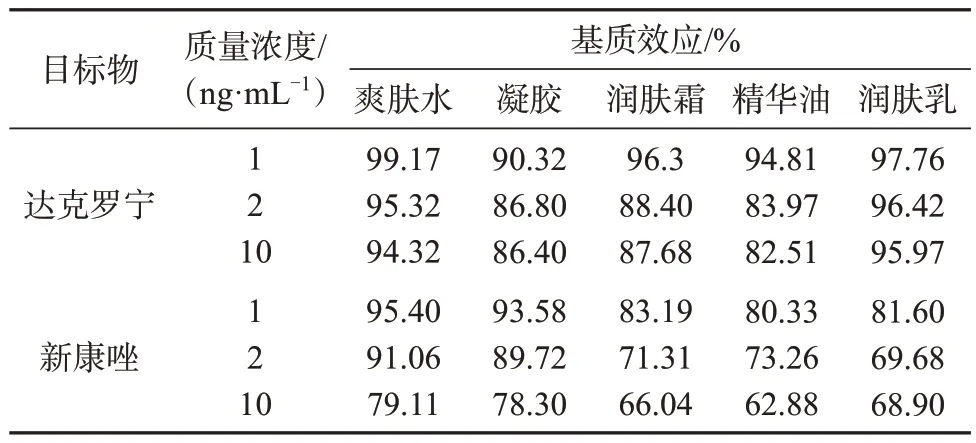
表6 空白化妆品样品中两种目标物基质效应Tab.6 Measurement results of matrix effects of two compounds to be tested in blank samples
2.6 标准工作曲线
在设定的分析条件下,测定系列浓度的目标物标准工作溶液和空白样品的目标物基质标准工作溶液,以目标物质量浓度(ρ)为横坐标,目标物色谱峰面积(Y)为纵坐标,进行线性回归(权重因子1/X),并绘制标准曲线、校准曲线,线性方程、相关系数、线性范围分别见表7 和表8。比较发现校准曲线线性明显优于标准曲线,故确定用校准曲线进行定量。

表7 标准曲线的线性方程、相关系数及线性范围Tab.7 Regression equation, correlation coefficient, and linear range of solution standard curve

表8 校准曲线的线性方程、相关系数及线性范围Tab.8 Regression equation, correlation coefficient, and linear range of blank matrix standard curve
2.7 检出限和定量限
以测定最低浓度点信噪比大于10 来确定方法定量限。结果显示,达克罗宁和新康唑溶液浓度为1 ng/mL 时,信噪比分别为1 406.1±5.02,364.1±8.28(n=6),空白样品加标0.1 μg/g 时,信噪比分别为3 313.0±11.25,277.3±8.77(n=6),均大于10。考虑不同仪器、不同色谱柱等因素,该方法中达克罗宁和新康唑的定量限均确定为1 ng/mL,当取样量为0.2 g时,可检测到的样品最低质量分数均确定为0.1 μg/g;按照3倍检出限为定量限原则,以上述定量限计算,确定该方法中达克罗宁和新康唑的检出限均为0.3 ng/mL,当取样量为0.2 g 时,可检测到的样品最低质量分数均确定为0.03 μg/g。
2.8 精密度和准确度
选择爽肤水、凝胶、润肤霜、精华油、润肤乳5种典型的空白化妆品样品作为验证基质,在方法定量限、两倍方法定量限和十倍方法定量限进行三水平空白基质加标试验,每个水平重复6次,进行重复性试验,结果见表9。
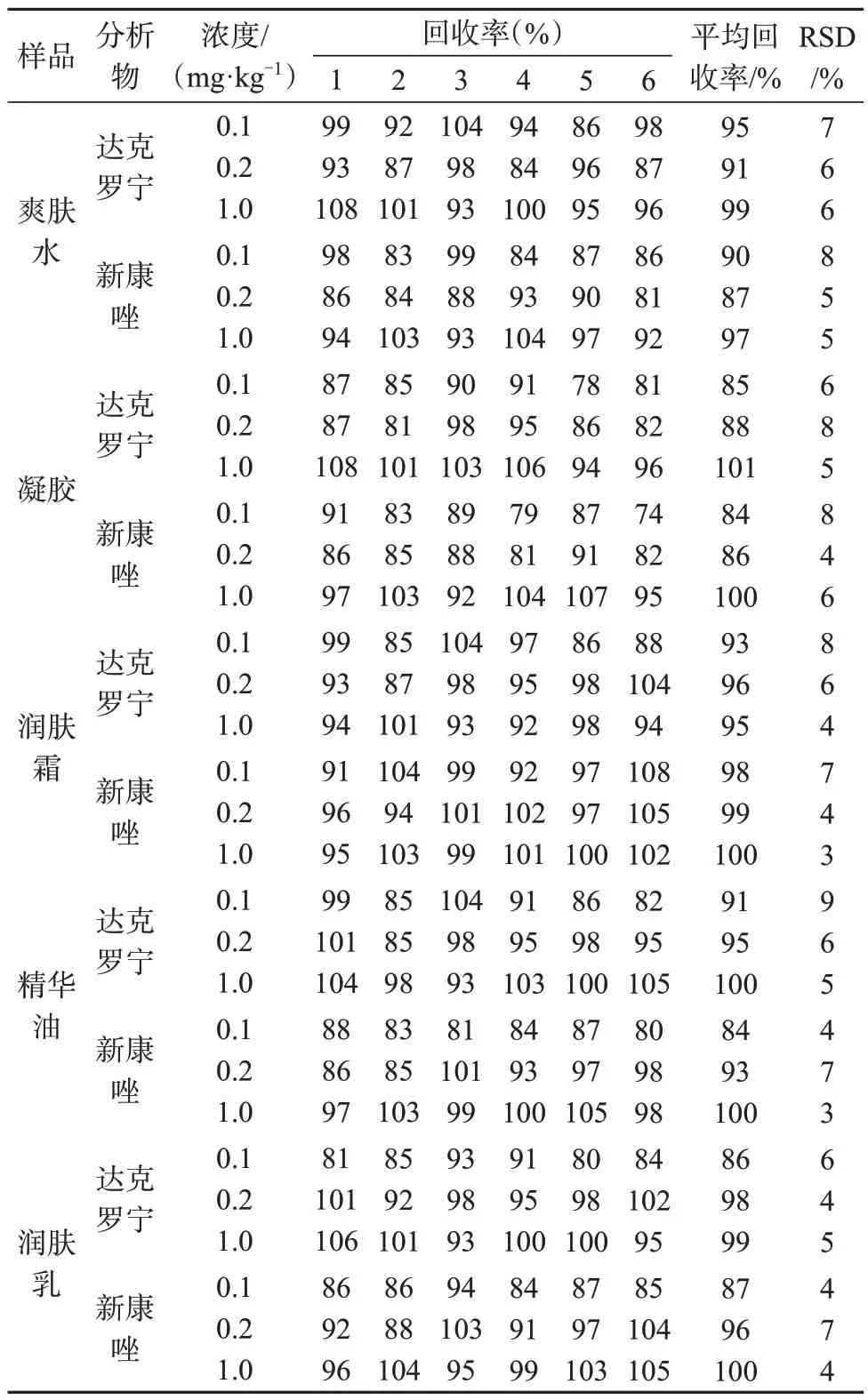
表9 5种典型基质中两种目标物的精密度和准确度Tab.9 Precision and accuracy assessment for two target substances in 5 typical matrices
根据化妆品补充检验方法管理办法,被测组分含量在0.01~0.1、0.1~1、1~10 mg/kg时,相对标准偏差分别为小于等于21%、小于等于15%、小于等于7.5%。被测组分添加水平小于等于0.1、0.1~1、1~100 mg/kg 时回收率范围分别为60%~120%、80%~110%、90%~110%。由表9 数据可知,该方法两种目标物的精密度和准确度均满足分析技术要求。
3 结语
通过对化妆品中两种目标物样品前处理方法的选择,分析条件的优化,最终建立了化妆品中达克罗宁和新康唑两种目标物的液相色谱-串联质谱检测方法。该方法分析速度快、灵敏度高、应用范围广,可以满足化妆品中达克罗宁和新康唑两种目标物的检测要求,为化妆品中非法添加的检测提供技术支持。
