异基因造血干细胞移植治疗儿童先天性角化不良3 例并文献复习
2024-03-16王英洁陈志伟买钰淼景沼贺董芃芃
王英洁 陈志伟 买钰淼 孙 盼 景沼贺 董芃芃 刘 健
1.郑州大学第一附属医院儿科(河南郑州 450052);2.河南省儿童医院儿内科(河南郑州 450052)
先天性角化不良症(dyskeratosis congenita,DKC),又称Zinsser‐Engman‐Cole 综合症,是一种罕见的遗传性骨髓衰竭综合征(inherited bone marrow failure syndromes,IBMFs),平均年发病率为1/1000000[1]。该病以骨髓衰竭和皮肤黏膜三联征(皮肤色素沉着、指甲营养不良、口腔白斑)为主要表现,67%的患者早期死于骨髓衰竭。造血干细胞移植(hematopoietic stem cell transplantation,HSCT)是纠正该疾病骨髓衰竭的唯一选择。本院自2019年6月至2022 年6 月对3 例DKC 患儿进行了HSCT 治疗,本文回顾性分析3例患儿临床资料及相关文献。
1 对象与方法
1.1 病例资料
2019年6月至2022年6月于郑州大学第一附属医院行HSCT的DKC患儿3例,其中男2例、女1例,发病中位年龄为4.8(3.2~8.7)岁,移植时中位年龄为9.0(4.8~11.4)岁。3例患儿中1例患儿(例3)就诊时未出现躯体症状,2 例(例1、例2)出现指甲萎缩及视网膜病变。3 例患儿入院后血常规均三系减低,骨髓细胞学检查提示再生障碍性贫血骨髓象。分别取先证者及其父母外周血2mL提取血液基因组DNA,对相关基因各外显子编码区域的序列突变情况进行检查,检测结果示:例1患儿TINF2基因错义杂合突变c.845 G>A:p.R 282 H;例2 患儿TINF 2基因存在1处杂合性突变c.845G>A:p.R282H;例3患儿RTEL 1基因存在一处杂合性突变c.1489 G>A:p.A497T,结合骨髓细胞学及细胞遗传学检查均可排除骨髓增生异常综合征(MDS)及急性髓系白血病(AML),治疗过程中3 例患儿均出现进行性骨髓衰竭,移植前产生输血依赖。
本研究经郑州大学第一附属医院伦理委员会批准(No.2023‐KY‐0254‐002),且监护人签署知情同意书。
根据目前国际推荐的DKC 诊断标准[2],符合以下1 条即可考虑诊断:①具有完整的皮肤黏膜异常三联征表现。②具有皮肤黏膜异常三联征表现之一,实验室检查出现骨髓衰竭,且具有其他DKC 临床表现。③再生障碍性贫血或MDS 或肺纤维病,存在DKC 相关致病基因阳性。④具有 Hoyeraal‐Hreidarsson 综合征的 4 条及以上临床表现:如生长发育迟缓、智力障碍、小颅畸形、骨髓衰竭、免疫功能缺陷和小脑发育不全。⑤端粒缩短(小于同年龄端粒长度第1百分位)及DKC 临床特点。3例患儿均可诊断DKC,治疗上移植前2例患儿口服环孢素,其中1例患儿同时口服司坦唑醇,1例患儿仅口服司坦唑醇,疗效差。见表1。
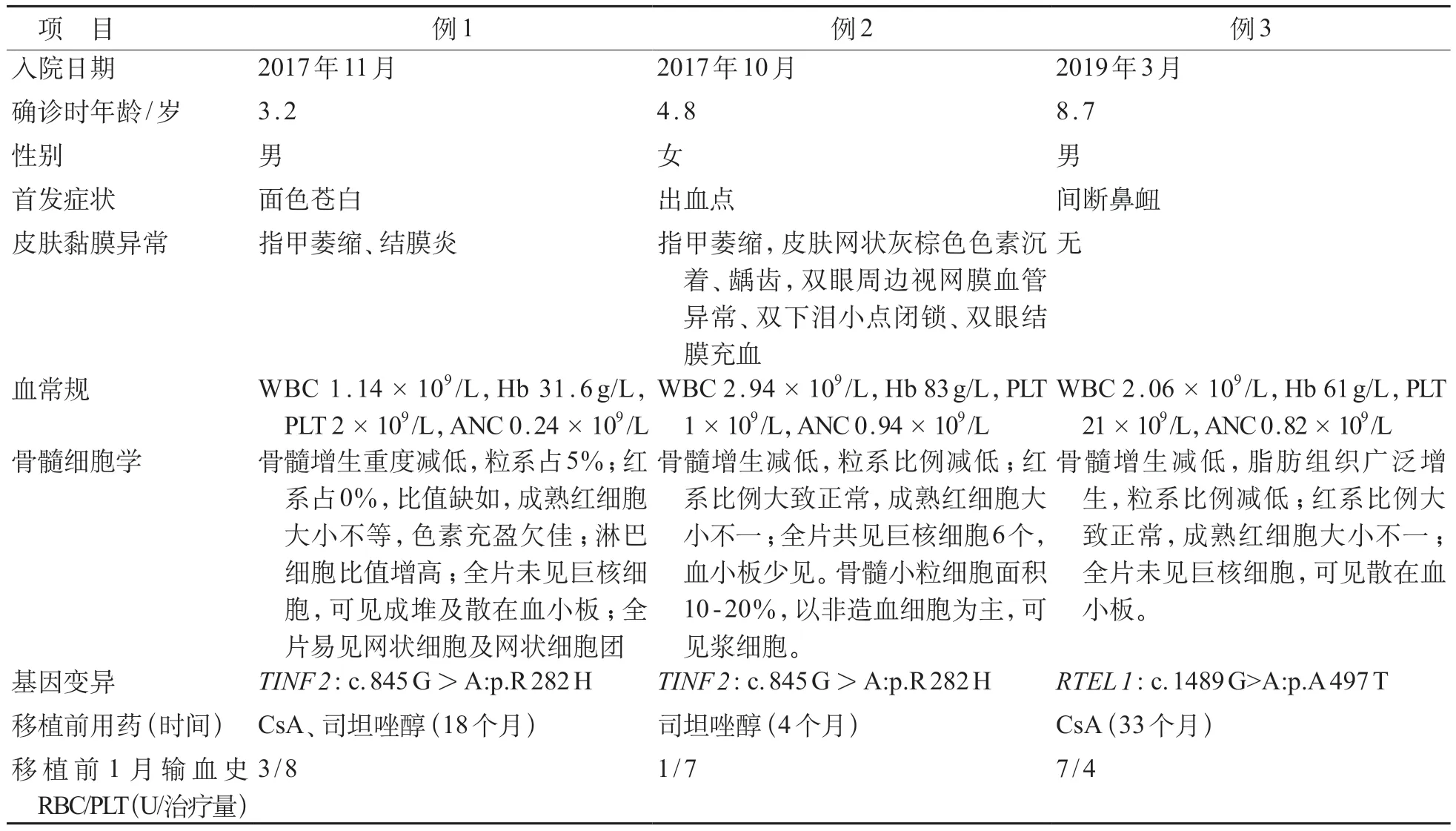
表1 3例接受造血干细胞移植治疗DKC患儿一般资料
1.2 文献复习
以“造血干细胞移植”和“先天性角化不良”为关键词检索万方和中国知网数据库(建库至2023年1 月)检索到2 篇文献;以“hematopoietic stem cell transplantation”和“Congenital Dyskeratosis”为关键词在PubMed 检索(建库至2022 年6 月)共检索到109 篇;通过阅读标题和摘要排除不相关的文献81篇,排除综述及个案报道19篇,最终纳入了9篇文献,共包含168 例HSCT 治疗DKC 的病例,男性121 例(72.02%),女性47例(27.98%)。
1.3 造血干细胞移植
患儿移植前评估达到移植指征[3]。供者选择:例1 为无关全相合供者(matched unrelated donor,MUD),例2 为无关9/10 相合供者(mismatched unrelated donor,MMUD),例3为不含相关基因变异的亲缘全相合供者(matched sibling donor,MSD)。供者情况、供受者血型及HLA 配型情况见表2。采用以氟达拉滨(Flu)为主的减低剂量预处理(RIC)方案:Flu 40 mg/(m2·d),d‐7~d‐4;环磷酰胺(CTX)20 mg/(kg·d),d‐5~d‐2;抗人胸腺淋巴细胞球蛋白(ATG)2.5 mg/(kg·d),d‐5~d‐2。

表2 供受者血型、HLA配型及干细胞输注情况
1.4 预防并发症
1.4.1 移植物抗宿主病 采用环孢素(cyclosporinA,CsA)、吗替麦考酚酯(MMF)和小剂量甲氨蝶呤(MTX)预防移植物抗宿主病(GVHD)。CsA 3~5 mg/ (kg·d),从-7 d 开始静脉滴注,待肠道功能恢复后改为口服,用药期间调整血药浓度达到150~250 ng/mL,如病情稳定,移植后半年开始逐渐减药至停药;MMF 从-7 d 按照30 mg/(kg·d)开始口服,连用30 d 酌情停用;移植后短程应用小剂量MTX,+1 d 予15 mg/m2,+3 d、+6 d 用10 mg/m2,可酌情在+11d加用10mg/m2加强GVHD预防。
1.4.2 肝静脉闭塞症 从-7 d开始应用前列地尔及熊去氧胆酸,出院后继续口服熊去氧胆酸至移植后+30d。
1.4.3 感染 预处理过程中给予更昔洛韦10 mg/(kg·d),移植后常规应用阿昔洛韦20mg/(kg·d)防治病毒感染,每周检测EBV‐DNA和HCMV‐DNA荧光定量PCR,根据结果调整用药。自预处理开始应用卡泊芬净预防真菌感染,移植后胃肠功能恢复后改为泊沙康唑口服制剂。
1.5 干细胞输注、支持治疗及植入评估
1.5.1 干细胞输注 3 例患儿中,例1、2 均为单次输注非血缘供者外周血干细胞(PBSC),例3 回输其兄PBSC;回输供者干细胞总数中,单个核细胞(MNC)中位计数为13.7(8.3~17.3)×108/kg,CD34+细胞中位计数为8.2(6.3~13.1)×106/kg。患儿自移植后+5 d或粒细胞<0.2×109/L开始应用粒细胞集落刺激因子(G‐CSF),及重组人血小板生成素刺激造血直到造血重建。
1.5.2 支持治疗 期间患儿血红蛋白低于60g/L或血小板低于20×109/L 即予以成分输血,输注的悬浮红细胞及机采血小板均经25Gy Co照射以减少移植后相关GVHD。
1.5.3 植入评估 连续3 天外周血中性粒细胞计数(ANC)>0.5×109/L 为粒细胞植入,在没有输血小板的情况下连续7 天外周血血小板>20×109/L为血小板植入。以荧光标记复合扩增短串联重复(STR‐PCR)检测植入情况。
1.6 随访
随访至2023 年1 月,随访中位时间19(11~42)个月。移植后100天内每周复诊1次,复诊复查血常规、血生化、铁蛋白、血药浓度、病毒载量等,定期复查骨髓象、免疫功能及嵌合率等,后根据患儿检查结果逐渐延长复查时间间隔,移植1年后每年评估1次生长发育并进行实体瘤筛查。
2 结果
2.1 预处理耐受
例1化疗耐受性可,例2预处理过程中出现药物性肝损害,给予保护肝脏药物治疗后转氨酶逐渐下降;使用ATG后出现发热、面部红疹及结膜炎,化疗结束后症状减轻;例3化疗后出现发热、腹泻,化疗停止并对症应用退热药物及蒙脱石散、益生菌后症状好转。
2.2 造血重建
3例患儿造血均顺利重建,中性粒细胞植入中位时间11(10~11)d;血小板植入中位时间11(10~13)d。3例移植嵌合率都为完全嵌合。
2.3 移植后合并症及处理
例1、例2 发生不同程度急性/慢性GVHD,按Thomas分度法和Glucksberg分度法,例1出现I级急性GVHD(皮肤Ⅰ度),例2出现Ⅱ级慢性GVHD(皮肤、口腔Ⅱ度,肝脏Ⅰ度),经联合免疫抑制治疗明显好转。
例1、例3患儿均合并BK病毒感染,通过抗病毒、输注丙种球蛋白后BK 病毒复制指数下降至<500拷贝/mL。
移植后2月,例1出现可逆性后部脑白质综合征(reversible posterior leukoencephalopathy syndrome,RPLS),移植后半年出现结膜炎,经对症处理后症状好转,例1 和例3 移植后出现出血性膀胱炎,伴BK病毒感染,予水化碱化尿液及抗病毒治疗后症状逐渐好转。
2.4 转归
随访至2023 年1 月,3 例患儿均存活,例1 指甲萎缩及结膜炎无明显改善,例2存在口腔粘膜白斑,口服免疫抑制剂可控制症状,躯体症状仍然存在,例3患儿无明显皮肤粘膜异常,供受者间基因嵌合率持续为完全供者型,骨髓恢复正常造血,且生长发育同正常同龄儿童,目前尚未发现合并实体肿瘤。
3 讨论
DKC是一种罕见的遗传性多系统疾病,男性患病率相对较高。目前认为DKC 是由编码端粒关键成分的基因变异引起的,是一种罕见的端粒功能障碍性疾病(telomere biology disorders,TBD),已知DKC致病相关基因共18种[9‐10],涉及X连锁隐性遗传、常染色体显性遗传及常染色体隐性遗传等多种不同的遗传方式。
DKC 常以皮肤黏膜三联征为主要临床表现,并伴有骨髓衰竭、肺纤维化、肝硬化等多种全身症状。但是,并非所有DKC 患者均伴有上述表现[6]。Revesz综合征是一种由于TINF2基因变异所致的罕见DKC,1992年被首次提出[7]。除典型DKC特征外,还表现为双侧渗出性视网膜病变、宫内生长迟缓、颅内钙化和神经认知缺陷。TINF 2基因变异是其发生原因。这些患者的端粒长度非常短,常在5岁前出现骨髓衰竭。本研究例1和例2均为TINF2基因变异,出现骨髓衰竭年龄分别为3.2和4.5岁,与既往研究相符。Hoyeraal‐Hreidarsson综合征是一种以小脑发育不全、骨髓功能衰竭、宫内生长受限和免疫缺陷为特征的DKC严重变异,多为RTEL1和DKC1基因异常,常导致儿童早期死亡[8]。本研究例3为RTEL1基因变异所致DKC,无明显躯体异常。
DKC 患者发生骨髓衰竭、MDS/白血病和其他恶性肿瘤的风险增加,骨髓衰竭通常是发病和死亡的主要原因[4‐5]。本研究中报道的3例患者均有进行性骨髓衰竭表现。例1最初因患儿面色苍白就诊,伴四肢指甲萎缩,治疗后期出现视网膜病变。例2 和例3分别以出血点和间断鼻衄为首要表现,例2在疾病发展过程中出现典型的皮肤粘膜三联征且伴有龋齿、双眼周边视网膜血管异常、双下泪小点闭锁等畸形体征,伴典型的眼部、皮肤黏膜症状。3 例患儿骨髓细胞学检查均为再生障碍性贫血骨髓象且有细胞遗传学改变,根据目前国际推荐诊断标准[2]可诊断为DKC。
DKC伴骨髓衰竭与获得性再生障碍性贫血的免疫介导发病机制完全不同,不能采用免疫抑制治疗,故例1和例3应用CsA免疫抑制治疗效果差。雄激素可通过增加促红细胞生成素或作用于促红细胞生成素受体促进造血,一项针对人类细胞系和再生障碍性贫血小鼠模型的研究表明,雄激素可以增加端粒酶表达,进而增加端粒长度[11],有效率可达到60%~70%,联合小剂量糖皮质激素也有一定效果,但长期应用有明显的不良反应。本研究3例患儿中例1和例2均使用了雄激素治疗,效果均不明显。
HSCT可使伴骨髓衰竭的DKC患者获得造血功能重建,也是目前唯一可以治愈该病的方法,3例患者均伴进行性骨髓衰竭并产生输血依赖,于我院行HSCT。预处理的选择上,DKC患者往往对清髓预处理(MAC)方案更敏感,大剂量的放化疗导致患儿预后较差,国际血液和骨髓移植研究中心的一项包含34例移植患者的相关研究证实,非亲缘供体移植清髓治疗增加患儿病死率,主要原因是肺部疾病晚期死亡[12]。在一项94例患者均接受RIC方案的研究可见,5年和10年总生存率(OS)分别为59%和30%,晚期死亡主要归因于肝和肺纤维化及其他血管并发症[13]。因此尽管DKC的理想预处理方案没有共识,目前常用以Flu 为基础的RIC 方案,避免或限制使用辐射和烷基化剂以减少后期影响[14‐16]。Bhoopalan等[17]报道了9例DKC患者在单中心接受以Flu为基础的RIC 方案后接受allo‐HSCT,OS 为66.7%,与Nelson等[16]报道的71%的OS相似,这些研究均证实了该方案的安全性及可行性,本研究3 例患儿均采用以Flu为基础的RIC方案,随访中位时间19个月,均存活。
慢性GVHD 与DKC 典型的皮肤、肝脏和肺受累不易区分,且慢性GVHD 影响患儿生存质量,例1、例2 均有不同程度的器官受累,与原发病症状不易区分,应用免疫抑制治疗后稍有好转,提示存在GVHD,考虑可能与例3 供者为MSD 有关。另有研究表明阿仑珠单抗比ATG更有利于降低慢性GVHD发生率[13],且更有利于降低移植后淋巴细胞增殖障碍的风险[18],但也因T 和B 淋巴细胞破坏引起了严重免疫抑制,进而增加感染风险[19]。但本研究3 例患儿均采用ATG,未出现严重GVHD,例2目前存在口腔粘膜白斑,不能排除局限性口腔慢性GVHD,局限性口腔慢性GVHD可能增加口腔鳞状细胞癌的发生风险[20],所以即使程度较轻,仍予积极处理,口服免疫抑制剂并局部应用地塞米松含漱液控制症状,定期进行致癌监测。
肺部疾病常被认为是DKC患者HSCT后死亡的主要原因。在最近的一份报告中,移植前肺功能异常的患者移植后发生重大肺部疾病的风险更高。在该研究中,移植后出现肺部并发症的中位时间为4.7年[21]。此外在一项大型队列研究中证实,与未接受HSCT的DKC患者相比,接受HSCT的DKC患者年轻时继发性癌症的累积发生率显著增加[22],目前3例患儿均未出现肺功能异常及恶性肿瘤出现,后续可继续监测随访并建议终身筛查。但HSCT 无法修复DNA及更新组织,DKC所致躯体异常无法改善,低剂量全反式维甲酸对患者的皮肤和指甲有一定效果,不良反应和长期影响尚不确定[23]。
综上所述,虽观察病例较少,但结果表明采用“Flu+低剂量CTX+ATG”预处理方案的可行性及安全性。GVHD 仍是影响患儿长期生存质量主要因素,且除实体器官移植外,目前尚无治疗肺纤维化、肝硬化的有效方法,由于这些并发症影响HSCT 后的生存,多学科合作对于早期发现肝肺问题至关重要。因此,探索个体化方案治疗DKC的临床研究非常必要。
