基于图注意力网络预测人类微生物与药物关联
2024-03-07史赛如
史赛如,孔 舒,张 冀
1. 河南科技大学数学与统计学院(河南洛阳 471023)
2. 北京建筑大学理学院(北京 102616)
人类微生物是一个复杂而多样的群落,对人类健康有重要影响。成人体内大约有1 014 个细菌,相当于人类细胞总数的10 倍,这些细菌可以产生大量的基因产物支持人体内的各种生化或代谢活动[1]。微生物对人类健康起着重要作用,可能导致转录组、蛋白质组和代谢组的变化,从而进一步损害人体组织,最终导致各种疾病,如肥胖、癌症和糖尿病等[2-4]。研究表明,微生物参与药物的吸收和代谢,从而调节药物的疗效和毒性[5-7]。因此,微生物-药物关联(microbe-drug association, MDA)研究受到越来越多的关注。
大量潜在的微生物-药物关系已被既往研究证实。Kovac 等证明了粪肠球菌和白色念珠菌菌株对环丙沙星轻微敏感[6]。Szczuka 等研究发现,环丙沙星可抑制表皮葡萄球菌生物膜的形成[7]。然而,传统的湿实验室实验用于揭示微生物与药物之间的关联是费力且昂贵的。因此,有效和准确地预测MDA 的计算方法是对实验方法的补充[8-11]。Sharma 等于2017 年开发了一种预测代谢酶和肠道细菌种类的计算方法,可用于药物分子生物转化[8]。Zhu 等提出了一种基于KATZ 测量的微生物-药物预测模型[10]。Long 提出了一种基于图卷积网络的MDA 预测框架(GCNMDA),该框架应用条件随机场可以确保相似的节点具有相似的表示[11]。虽然已经提出了许多预测MDA的计算方法,但在特征提取过程中无法保留微生物和药物的综合特征。为了解决上述问题,本研究基于图注意力网络(graph attention network,GAT)提出了一种新模型GATMDA,用于二分网络中的MDA 预测。
1 资料与方法
1.1 数据来源
本研究数据来源于三个已知微生物与药物关联的数据集[11]。第一个数据集是MDAD 数据集,去除冗余信息后,包含1 373 种药物和173 种微生物之间的2 470 种已知关联;第二个数据集是aBiofilm,存储了抗生物膜制剂的资源及其在抗生素耐药性方面的潜在影响,其中挑选了2 884 种微生物-药物对进行研究;第三个数据集是Drug Virus,记录了多种人类病毒的相关化合物的活性及其发展,包含95 种病毒和175 种药物间的933种关联。上述三个数据集的详细信息见表1。

表1 三个数据集的详细数据Table 1. Detailed data for three datasets
1.2 方法
1.2.1 人类微生物与药物的关联
为了推测微生物-药物网络中的新关联,本研究将问题框架视为一个生物二分网络的关联预测任务。在此网络中,微生物和药物分别被表示为两类不同的节点。定义药物Nd的节点集为A= a1,a2,… ,aNd,微生物Nm的节点集则被定义为B= b1,b2,… ,bNm。网络中的边是微生物与药物之间的关联,可以表示为邻接矩阵 Y ∈ RNd×Nm 。当Yi,j=1 时,表示一个微生物bj(1 ≤j≤Nm)对应一个药物ai(1 ≤i≤Nd)。相反,Yi,j=0 表示关联未知。研究目标是生成一个与Y相同维度的预测矩阵F*来预测未知的关联。图1 展示了GATMDA 算法流程。

图1 GATMDA算法的原理示意图Figure 1. The schematic diagram of the GATMDA algorithm
1.2.2 构建异构网络为了将网络信息纳入数据整合,根据邻接矩阵Y构建了一个包括微生物网络Sm、药物网络Sd和MDA 网络的异构网络[12]:
1.2.3 图注意力网络
GAT 是一个基于空间的图卷积网络,核心在于聚合邻居特征的过程中聚焦更多重要邻居的特征贡献[13-14]。GAT 在本研究中被用于提取微生物和药物特征。具体而言,对于上面定义的二进制网络的邻接矩阵,GAT 定义如下:
其中 H(l)是节点的l层嵌入,l=1, ...,L,σ (.) 是非线性激活函数 (ReLU),GAT 表示单个图注意力层,整个L层GAT 架构由多个图注意力层堆叠。初始输入是一组节点特征其中n是节点的数量,F是每个节点中特征的数量。该层生成一组新的节点特征并且通过将权重矩阵 W ∈ RF'×F应用于每个节点。注意力系数为:
在通过softmax 函数进行归一化之后,将系数变为:
将式3 代入式4,可以表示注意力机制的系数如下:
其中,a是注意力系数,表示参数化权重向量,LeakyReLU 表示激活函数,T表示矩阵转置,| | 是连接运算,Ni是节点i的邻居集合。在计算归一化注意力系数后,每个节点的最终输出特征可以计算为:
对于第一层构造初始嵌入H(0),如下所示:
1.2.4 多核融合
多层GAT 模型可以计算表示具有不同图结构的信息的多个嵌入。由于不同的嵌入表示不同的结构信息,因此由不同嵌入组成的核将表示不同角度节点之间的相似性。结合现有的相似性矩阵,可以得到药物空间 SD= Sd, Kh1d, … , KhLd和微生物空间 SM= Sm, Kh1m, … , KhLm的核集。 Khld和 Khlm分别是药物和微生物嵌入的核矩阵。为了提高预测性能,分别在两个空间中对上述核进行了多核融合,通过加权方法组合多个核矩阵。组合内核定义如下:
其中Sid和Sim是药物和微生物核集中的第i个核,ai和bi是每个核对应的权值,L是对应的层数。
1.2.5 解码器
最后,通过应用一个改进的对偶拉普拉斯正则化最小二乘(DLapRLS)框架来预测关联,提高预测性能。DLapRLS 是基于两个特征空间的核矩阵模型。在这项工作中,基于DLapRLS,将药物-微生物特征空间组合纳入最小二乘框架来构建一个新的目标函数。目标函数的定义如下:
DLapRLS 方法的目的是通过最小化以上目标函数寻找最优的预测结果。其中,‖.‖F是 Frobenius范数,Ytrain∈RNd×Nm是训练集中的MDA 的邻接矩阵;Kd∈RNd×Nd是可训练矩阵;αd,αmT∈RNd×Nm和Km∈RNm×Nm分别是两个特征空间中的融合核即微生物和药物之间的相似性度量。Kdad代表在药物空间下,药物与微生物的关联预测结果;Kmam代表微生物空间下,药物与微生物的关联预测结果。φ是用来平衡正则化项的衰减因子(Decay factor)。
由上述可得,Kdad和Kmam可以实现不同空间下的微生物和药物关联性的表示。故对两个空间的预测结果进行平均融合作为最终预测结果,进而有效整合微生物-药物的空间信息。因此,基于DLapRLS,来自两个特征空间的MDA 的最终预测F*组合如下:
上述F*也作为模型GATMDA 的最终输出结果,根据F*可以得到微生物和药物的关联预测矩阵,基于该矩阵的得分可以衡量微生物-药物关联性。
1.3 统计分析
采用Python 3.7 软件进行数据分析。本研究建立GATMDA 模型以预测微生物与药物的关联性,K 折交叉验证用于评估预测性能。在交叉验证期间,所有关联平均分为K 个部分。在每一次折叠中,选择其中一个作为测试集,其余用作训练集,用于训练和验证模型,总共 K 个折叠。模型的性能评估采用受试者工作特征(receiver operating characteristic, ROC)曲线和精确率-召回率(precision-recall, PR)曲线,评价指标为ROC 曲线下面积(area under the curve, AUC)和精确率-召回率曲线下的面积(area under the precision-recall curve, AUPR)。GATMDA 中重要的参数包括衰减因子(decay factor)φ、迭代时间(iteration time)N、学习率(learning rate)。
2 结果
2.1 参数敏感性分析
首先,在MDAD 数据集上使用5 折交叉验证(5-fold cross-validation, 5-CV)选择模型参数,设置默认参数层数L=3,嵌入维度分别为K1=256,K2=64,K3=32。衰减因子φ被用于调节公式(10)中正则化项的影响,φ的取值范围为0.000 005 至0.5,步长设定为10。如图2-A 和图2-D 所示,参数φ对模型性能的影响较为有限,表明模型具备一定的鲁棒性。当φ值为0.000 5时,模型达到最优性能。学习率也是一个非常重要的参数,当学习率太大时,模型很难收敛,较小的学习率可能会导致一个较长的训练过程,一个合理的学习率可以使模型收敛到局部最小值。因此,学习率被设定在{1e-1, 1e-2, 1e-3, 5e-3,1e-4, 5e-4},并对GATMDA 在各个学习率下的性能进行了评估。如图2-B 和图2-E 所示,学习率1e-1 从提升至5e-4,GATMDA 的性能先提高后略有下降,当学习率为1e-3 时,模型展现出最佳效果。迭代次数对模型同样至关重要,其决定了可训练参数的更新频率。本研究将N 的取值范围设定为1 至12,以1 为步长。图2-C 和图2-F分别显示了不同迭代次数下的AUC 值和AUPR值,当迭代次数为5 时,AUPR 值开始趋于稳定。为了使模型完全收敛,确定迭代次数为10。
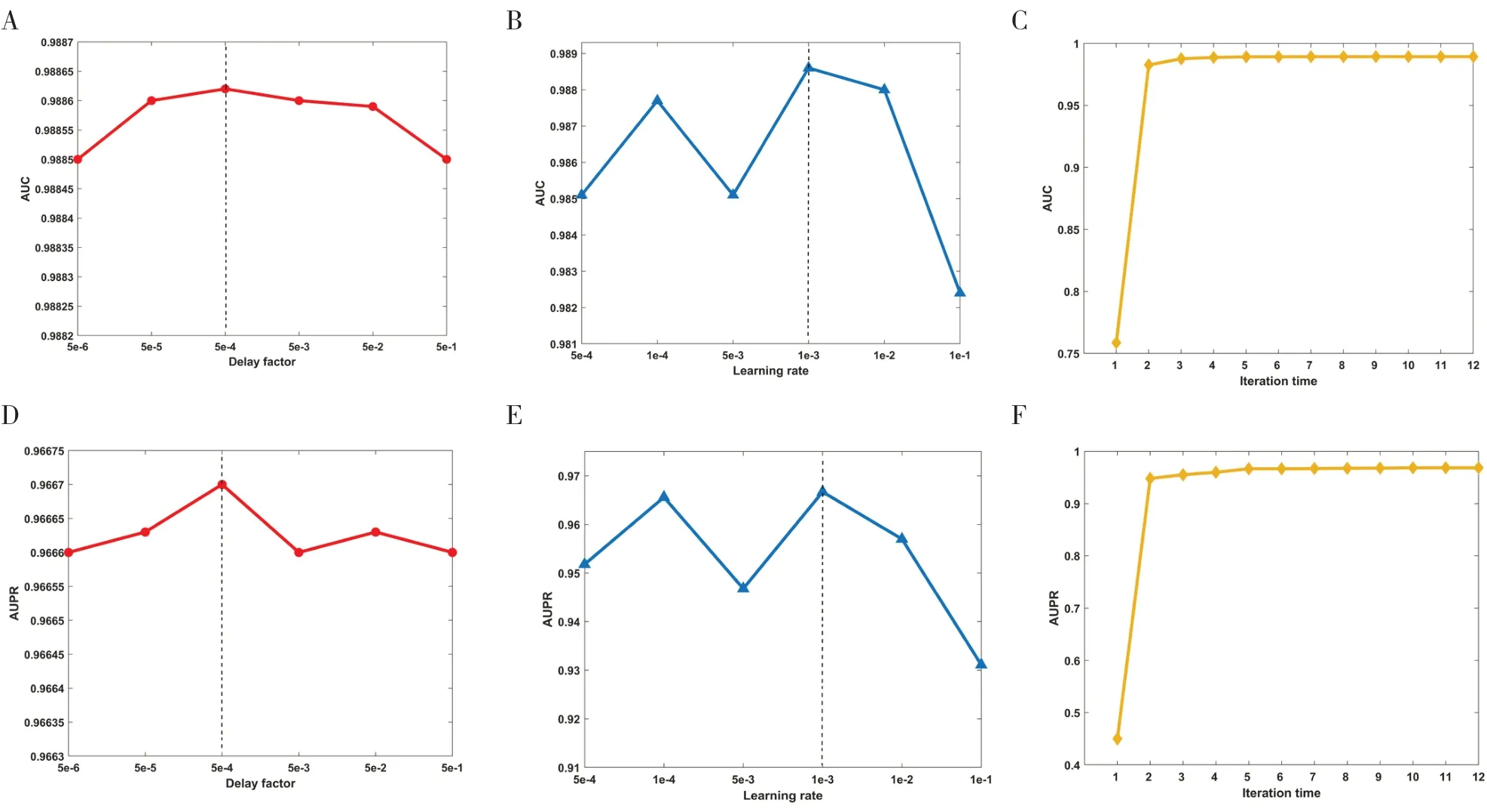
图2 GATMDA的参数灵敏度Figure 2. Parameter sensitivity of GATMDA
2.2 与现有预测方法的比较
将GATMDA 与现有的8 种生物二分网络预测方法进行比较,如表2 所示,在MDAD 数据集上,GATMDA 模型的预测性能最好(AUC=0.988 6,AUPR=0.966 7),优于其他8 个模型;在aBiofilm数据集上,GATMDA 模型的AUC 值和AUPR 值均最高(AUC= 0.994 1,AUPR=0.986 9);在Drug Virus 数据集上 ,GATMDA 模型的AUC 值最高(AUC=0.983 6),AUPR 值排名第二(AUPR=0.879 5)。
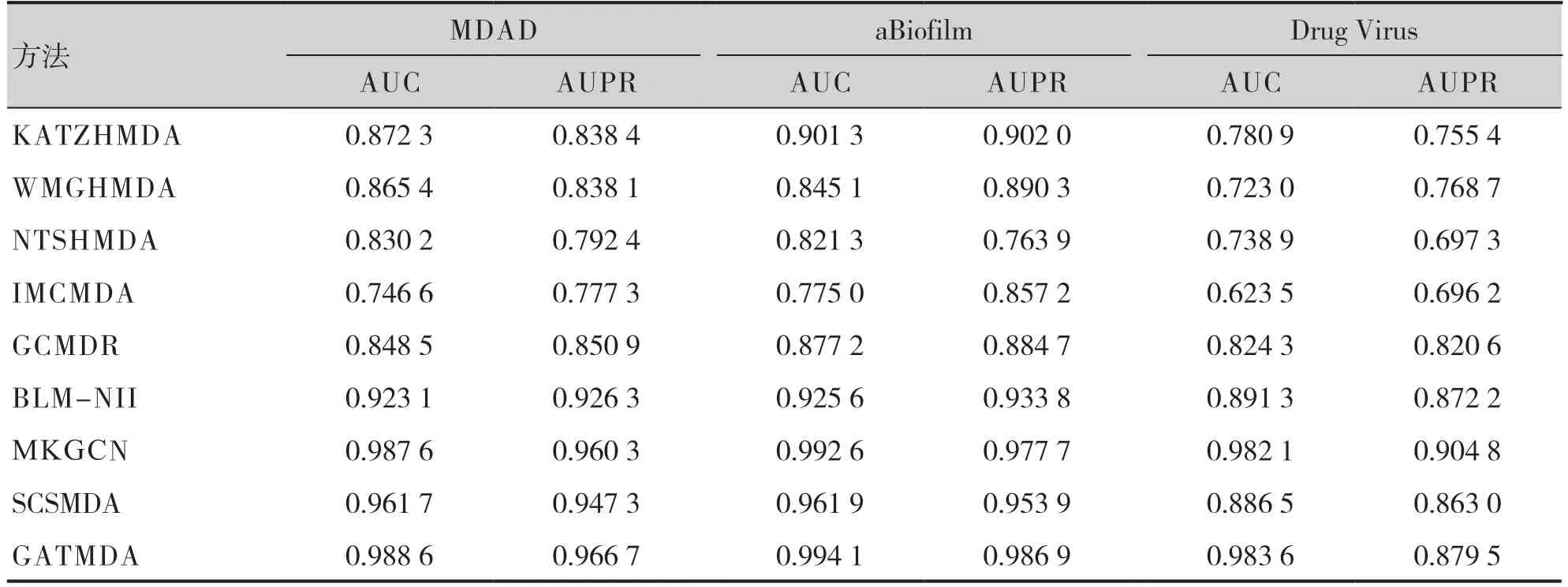
表2 三个数据集上不同预测方法在5折交叉验证下的性能比较Table 2. Performance comparison between different prediction methods on three datasets under 5-CV
本研究在2 折交叉验证(2-fold crossvalidation, 2-CV) 和10 折交叉验证(10-fold cross-validation, 10-CV)的设置下在三个数据集上对所有方法进行了比较,结果见表3 和表4。在2-CV 的条件下,GATMDA 模型在MDAD 数据集(AUC=0.982 6,AUPR=0.948 2)、aBiofilm数据集(AUC=0.984 1,AUPR=0.957 1)和Drug Virus 数据集(AUC=0.955 0)上展现出优于其他方法的预测性能。在10-CV 下,GATMDA 模型展示了最佳的预测性能,在MDAD 数据集上AUC值为0.989 3、AUPR 值为0.968 5;在aBiofilm 数据集上AUC 值为0.996 3、AUPR 值为0.984 4;在Drug Virus 数据集上AUC 值为0.986 3、AUPR值为0.904 0。GATMDA 在前两个数据集上超越了其他8 种评估方法,证明GATMDA 是一个预测MDA 的高效且强大的模型。GATMDA 模型基于3 种交叉验证方法在三个数据集上的ROC 曲线和PR 曲线见图3。
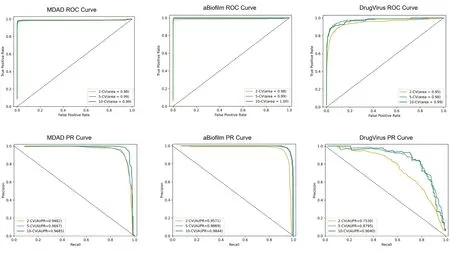
图3 GATMDA在MDAD、aBiofilm和Drug Virus数据集上的ROC和PR曲线Figure 3. The ROC and PR curves of GATMDA on the MDAD, aBiofilm and Drug Virus datasets

表3 三个数据集上不同预测方法在2折交叉验证下的性能比较Table 3. Performance comparison between different prediction methods on three datasets under 2-CV
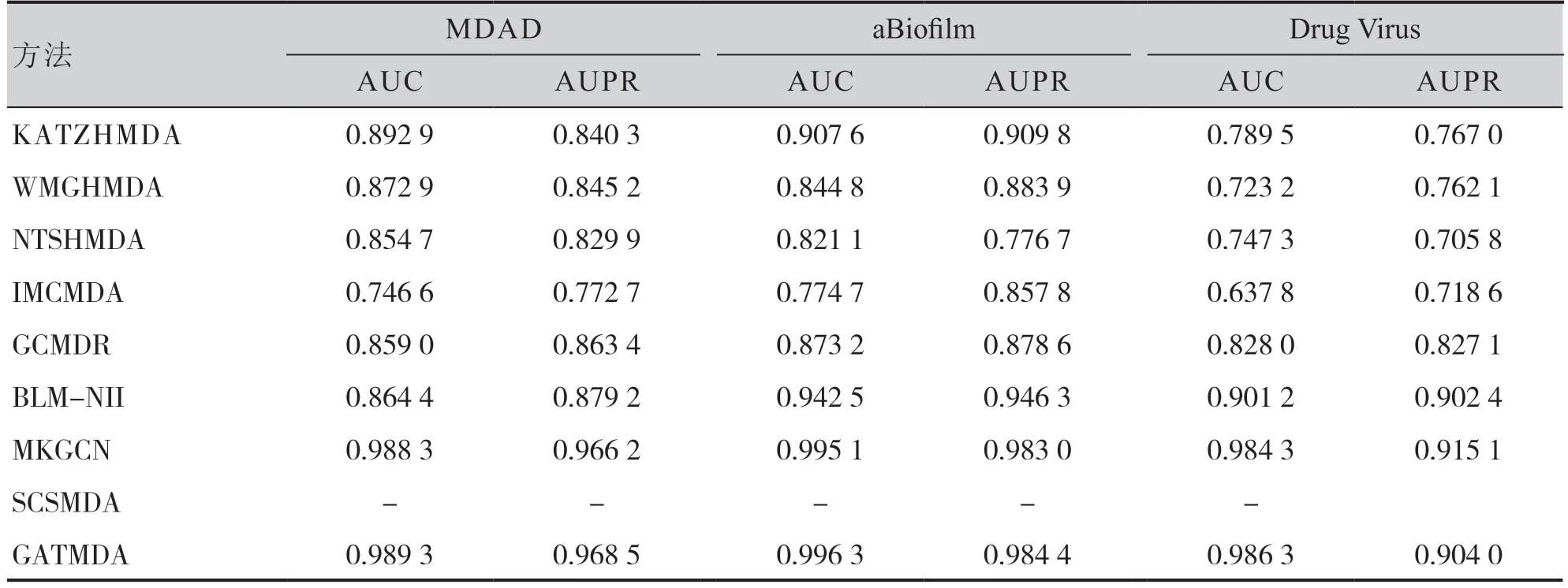
表4 三个数据集上不同预测方法在10折交叉验证下的性能比较Table 4. Performance comparison between different prediction methods on three datasets under 10-CV
2.3 案例研究
通过Drug Virus 数据集的案例研究,进一步测试GATMDA 的预测效果。案例研究选择HIV-1 测试模型的预测性能,并预测了可能有效治疗的药物。HIV 是一种逆转录病毒,可破坏CD4 T 细胞,是获得性免疫缺陷综合征的病原体。艾滋病毒分为两种类型:HIV-1,引起全球流行病;HIV-2,致病性较弱,主要局限于西非。因此,选择HIV-1 为案例进行试验。在试验中,Griffith 等测量了15 名HIV 感染患者中stavudine(2',3'-didehydro-3'-deoxythymine) 对HIV-1 的抗病毒功效,试验结果显示,stavudine 具有显著而持久的抗病毒作用[19]。Enfuvirtide 是一种新型HIV-1 融合抑制剂,在体外和体内均具有针对HIV-1 的有效抗病毒活性[20]。如表5 所示,预测HIV-1 相关药物中,前10 名药物全部在文献中得到支持;在预测的前20 种和30 种药物中,95%和93%的药物得到了文献的支持,并被证明可以治疗或预防HIV-1。选择175 种预测药物中和HIV-1 关联的前30 种药物测试GATMDA 的有效性,见图4-A。通过绘制条形图和散点图可视化前30 种预测HIV-1 的药物,见图4-B 和图4-C,这些预测结果证明了GATMDA 模型预测微生物-药物网络中潜在关联的能力。
3 讨论
居住在人体上的微生物在人类健康中起着关键作用[21]。预测MDA 可以促进个性化药物的有效开发,并了解微生物和药物之间的联系。与传统方法相比,计算方法能够在全球范围内识别靶向现有药物或针对具有已知微生物的新药的靶向微生物[22]。值得注意的是,MDA 预测也是生物二分网络中的一个链接预测问题[23-24]。本研究提出了一个计算框架GATMDA,用于预测微生物-药物的关联。GATMDA 由两部分组成,第一部分使用GAT 进行特征提取,实验表明,利用该机制可以生成更可靠的推理信息;另一部分是利用改进的DLapRLS 进行预测,充分利用了微生物-药物空间的信息进行预测。与传统的多核学习不同,本研究通过多层GAT 提取各种嵌入特征来构建核矩阵,可以提供不同的核矩阵,并实现使用多种信息的目的[25]。与现有的生物二分网络检测模型相比, GATMDA 模型在三个MDA 数据集上表现出了较好的预测性能。此外,关于HIV-1 的案例研究表明,GATMDA 可以准确地发现新的MDA。
虽然GATMDA 具有良好的预测性能,但对于不同密度的数据集仍存在一定的偏差。如GATMDA 在Drug Virus 数据集上的表现弱于在MDAD 和aBiofilm 数据集,这表明GATMDA 模型的泛化性能仍有改进的空间。微生物对药物治疗过程的影响包括激活、钝化和毒性,准确识别药物上未知微生物的类型是药物开发和精准医学的基本要求,但GATMDA 无法预测微生物-药物的类型。因此,为了更准确地了解微生物在药物治疗过程中的作用机制,建立一个有效的深度学习模型来预测微生物与药物之间的关系有待进一步研究。
综上,本研究通过构建GATMDA 模型,重点探讨了人类MDA 预测。GATMDA 模型侧重于结合GAT 和多核融合来探索微生物-药物的空间信息,进而有效地整合微生物与药物之间丰富的生物学信息,该模型具有捕捉微生物和药物之间复杂关联的能力,为预测微生物与药物关联提供了有效的新方法。
