5-氨基咪唑类化合物的制备及其活性研究
2024-03-05陈彩萍章潮军王小平徐作武徐慧婷
陈彩萍,章潮军,王小平,徐作武,徐慧婷
(1.浙江昌海制药有限公司,浙江 绍兴 312000;2.浙江可明生物医药有限公司,浙江 绍兴 312000;3.浙江绍兴文理学院 化学化工学院,浙江 绍兴 312000;4.脂溶性维生素浙江省工程研究中心,浙江 绍兴 312000)
含氮杂环化合物种类繁多,数量庞大,在自然界中广泛分布,许多天然含氮杂环化合物在动植物体内起着重要的生理作用[1-3]。5-氨基咪唑是一类具有多种生物活性的杂环类化合物[4-9],广泛用于抗血小板、抗菌、抗炎、抗癌等药物的研发,在医药领域具有重要的应用价值,同时它还是合成药物活性分子和天然生物活性分子的重要合成砌块,如被用于5-氨基咪唑核苷酸的生物合成,咪唑啉杀虫剂的合成,促性腺激素释放激素受体拮抗剂的合成等。5-氨基咪唑类化合物在药理学方面表现出广泛的生物活性[10-11],如抗癌活性,在脂肪细胞中通过WNT/B-连环蛋白通路抑制脂肪生成,通过AMP-活化的蛋白激酶依赖路径诱导细胞色素P450 4F2的表达和在骨骼肌中增加去乙酰化酶的表达。
5-氨基咪唑在化学和生物方面具有相当大的功能性,但是,传统的合成5-氨基咪唑的方法[12-13]都存在着或多或少的缺陷,大都存在着原子经济性和步骤经济性差、产率低、反应条件相对苛刻以及副产物多、后处理困难等不足,不适于大规模的工业生产,因此开发一种新的绿色、高效的合成5-氨基咪唑的方法就显得尤为重要。
目前,文献报道的5-氨基咪唑类化合物的合成方法主要有以下5 种:①硝基还原或者叠氮基还原法;②1-氨基-1,2,4-三氮唑法;③微波合成法;④原乙(甲)酸三乙酸酯法;⑤异腈法。
利用还原法[14-15]合成5-氨基咪唑的主要原料是5-硝基咪唑,或者5-叠氮基咪唑,通过使用Pd/H2催化剂对反应进行催化还原,从而得到所需要的5-氨基咪唑,反应方程式如图1所示。由于所使用的原料为5-硝基咪唑或5-叠氮基咪唑,原料来源不便,且使用Pd/H2催化,成本较高,不适于大规模的工业生产,而且H2易燃,在反应过程中极易引发爆炸,存在安全隐患。此外,使用5-叠氮基咪唑还原法制备5-氨基咪唑时,产率较低,仅有71%。
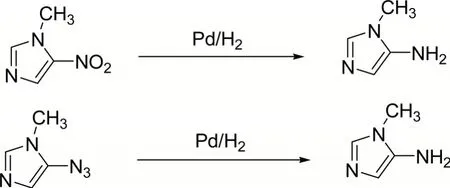
图1 还原法合成5-氨基咪唑的反应式
Donskaya O V 课题组[16]利用该方法合成5-氨基咪唑的主要原料是4-硝基咪唑和1-氨基-1,2,4-三氮唑,使用强碱叔丁醇钾为催化剂进行反应,反应方程式如图2所示。虽然该方法也可以成功合成5-氨基咪唑,且原料相对而言较为廉价,反应过程也比较安全,但是还是存在一定的问题,如产率太低,仅有39%,因此也不适于大规模的工业生产。
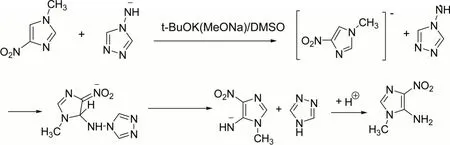
图2 1-氨基-1,2,4-三氮唑法合成5-氨基咪唑的反应式
Soh C.H.等[17]首先使用腈类化合物和原硫酸二乙酯在微波条件下80℃高收率合成硫代酰胺1,接下来在氯仿中80℃与苄溴反应,生成硫代亚胺类化合物2,然后再与氨基乙腈类化合物反应生成5-氨基咪唑化合物3,反应方程式如图3 所示。该方法与传统的合成方法相比,反应时间大大缩短,整个反应过程仅需25 min。但是由于该方法整个过程是在微波条件下进行,存在安全隐患。
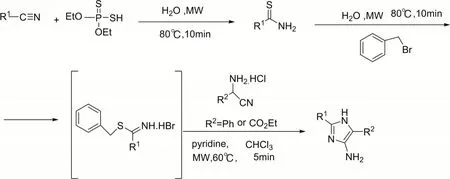
图3 微波法合成5-氨基咪唑的反应式
Ray S.与Das A.等[18]把2-氨基-2-氰基乙酰胺在干燥蒸馏的乙腈中加入原甲酸三乙酯,加热回流一段时间后冷却到室温时,加入伯胺进行反应,在容器表面会产生沉淀,将此产物用乙腈洗涤后得到粗产物,该法仅有一步实验流程,较为快捷,但是整个反应过程需要长时间的加热回流,产物沉淀之后,也需要用乙腈多次冲洗,后续操作过于麻烦,反应方程式如图4所示。
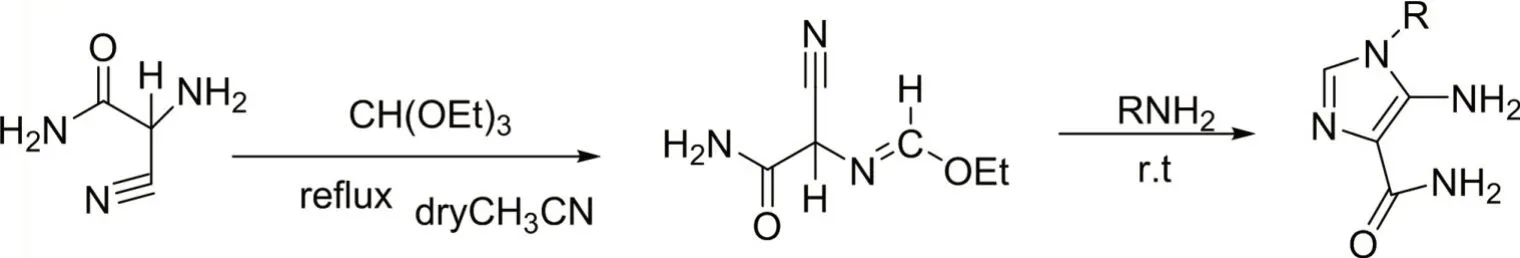
图4 原乙(甲)酸三乙酸酯法合成5-氨基咪唑的反应式
John T.Hunt 等[19]使用异硫脲1 和异氰乙酸乙酯钾盐2 在六甲基磷酰三胺中混合,以氯化亚铜为催化剂。在室温下反应几小时得到所要产物,但是在实验过程中,总是会产生相当多碳二亚胺的中间产物,比如,反应物2在标准条件下进行反应,实验后所得的产物产率只有30%,而且反应生成两种产物,选择性较差。因此,此法虽然能在常温下,利用一个合成步骤合成产物,但是产率极低,选择性较差,反应方程式如图5所示。

图5 异氰法合成5-氨基咪唑的反应式
本文提供一种一锅法制备5-氨基咪唑化合物的方法,合成系列化合物并通过MTT 比色法对其生物活性进行检测。合成路线如图6:以芳基硫脲和异氰为起始原料,DUB 为催化剂,制备5-氨基咪唑化合物,并对其进行表征分析。

图6 5-氨基咪唑化合物的制备路线
1 实验部分
1.1 仪器
核磁共振仪,AVANCE DMX ⅡⅠ400M,TMS内标,Bruker 公司;熔点仪,MP30,梅特勒;高分辨质谱仪(HRMS),岛津。
1.2 试剂(表1)

表1 试剂
1.3 5-氨基咪唑化合物的制备
于25 mL 的圆底烧瓶中加入磁力搅拌子,芳基硫脲(1.2 mmol),异腈(1.0 mmol)和DMF(5 mL),随后向上述体系中缓慢加入DBU(2.0 mmol),于室温下搅拌反应24 h,点板跟踪直至反应完全。反应结束后,往反应体系中加入饱和食盐水溶液和乙酸乙酯进行萃取,取有机层,反复萃取3次,合并有机层,用无水硫酸钠干燥,减压蒸馏得到粗品,再经柱色谱分离纯化(乙酸乙酯∶石油醚=1∶10)得目标5-氨基咪唑衍生物,详见表2。
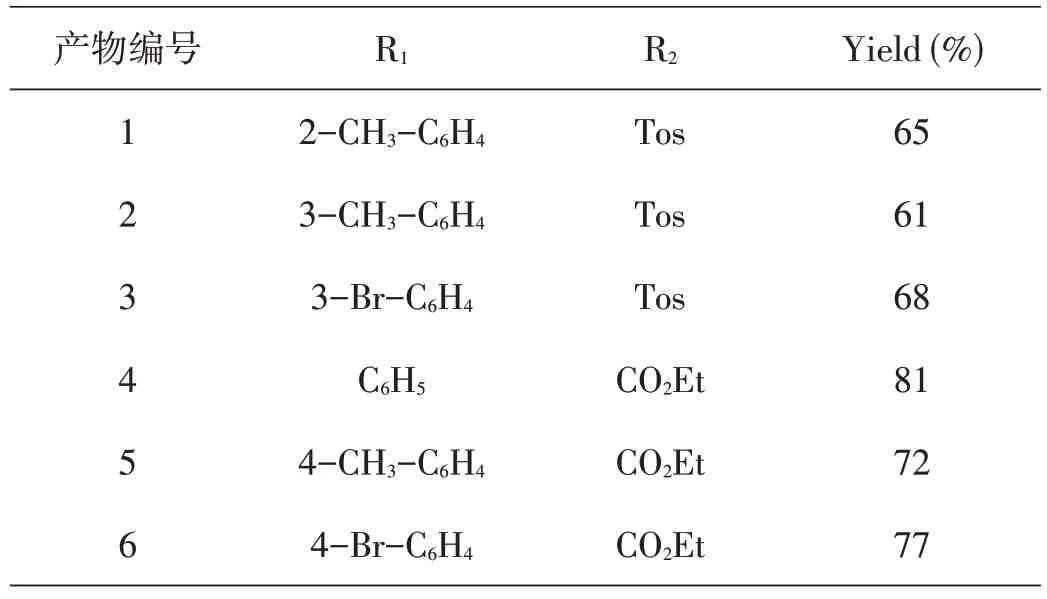
表2 产物及其收率
产物的结构为:
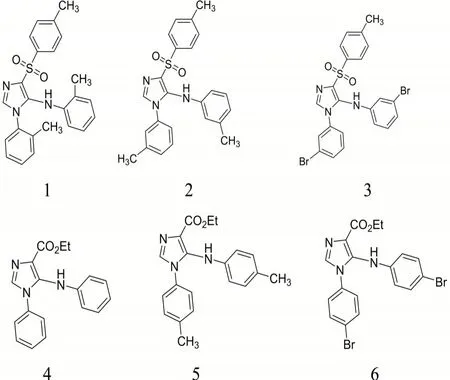
化合物1表征数据:产品为白色固体,收率65%,熔点198.6℃~200.9℃。1HNMR(500 MHz,CDCl3): δ 7.87(d,J = 8.5 Hz,2H),7.28-7.27(m,2H),7.14-7.09(m,2H),7.01-6.95(m,2H),6.92-6.91(m,1H),6.81-6.75(m,2H),6.66(s,1H),6.42(d,J=7.0 Hz,1H),2.40(s,3H),2.13(s,3H),2.10(s,3H).13C NMR(125 MHz,CDCl3): δ 143.94,140.12,139.16,138.92,134.85,134.74,133.27,130.99,130.44,129.77,129.51,127.53,127.25,126.58,126.12,125.61,123.62,119.95,21.62,17.78,17.57。
HRMS(ESI): m/z calcd for C24H24N3O2S [M+H]+:418.158 4,found: 418.158 5。
化合物2表征数据:产品为浅黄色固体,收率60%,熔点200.8℃~203.3℃。
1HNMR(500 MHz,CDCl3): δ 7.81(d,J = 8.5 Hz,2H),7.48(s,1H),7.23(d,J=8.0 Hz,2H),7.17-7.13(m,3H),6.91-7.86(m,2H),6.61(d,J = 7.5 Hz,1H),6.38(d,J=7.5 Hz,1H),6.34(s,1H),2.39(s,3H),2.25(s,3H),2.09(s,3H).13C NMR(125 MHz,CDCl3) δ 144.06,142.02,139.72,138.81,138.75,136.58, 134.82, 134.77, 129.80, 129.51, 129.31,128.82, 127.66, 127.59, 124.94, 123.06, 121.29,118.75,115.11,21.71,21.30,21.29。
HRMS(ESI): m/z calcd for C24H24N3O2S [M+H]+:418.158 4,found: 418.158 0。
化合物3表征数据:产品为黄色固体,收率69%,熔点195.6℃~197.6℃。
1HNMR(500 MHz,CDCl3): δ 7.85(d,J = 8.5 Hz,2H),7.72(s,1H),7.55(d,J=8.5 Hz,2H),7.53(s,1H),7.34-7.32(m,2H),6.93-6.88(m,3H),6.70(s,1H),6.69-6.65(m,1H),6.45(d,J = 8.0 Hz,1H),2.38(s,3H).13C NMR(125 MHz,CDCl3)δ 144.90,140.11,138.76,135.31,134.98,133.69,133.06,132.58,131.04,129.85,129.38,128.35,128.21,127.99,123.66,120.54,117.52,113.48,21.66。
HRMS(ESI): m/z calcd for C22H17Br2N3O2S [M+H]+:544.940 8,found: 544.940 3。
化合物4 表征数据:产品为浅黄色固体,收率79%,熔点145.0℃~147.6℃。
1H NMR(CDCl3,500 MHz)δ: 7.41-7.37(m,3H),7.28-7.26(m,2H),7.21(d,J=8.5 Hz,2H),7.11(d,J = 8.0 Hz,2H),6.98-6.78(m,2H),6.07(s,1H),4.43-4.38(m,2H),1.43-1.40(m,3H);13C NMR(CDCl3,125 MHz) δ: 175.36,150.70,144.22,142.62,138.59,136.09,129.32,129.00,127.59,124.27,122.09,117.62,63.24,14.75。
HRMS(ESI): m/z calcd for C18H17N3O2[M+H]+:307.132 1,found: 307.131 5。
化合物5 表征数据:产品为白色固体,收率73%,熔点148.0℃~149.1℃。
1H NMR(CDCl3,500 MHz)δ:7.41(s,1H),7.14(d,J=8.5 Hz,2H),7.06(d,J=8.5 Hz,2H),6.80(d,J=8.0 Hz,2H),6.69(d,J = 8.0 Hz,2H),6.0(s,1H),4.39(q,J=8.0 Hz,2H),2.37(s,3H),2.26(s,3H),1.39(q,J=8.0 Hz,3H).13C NMR(CDCl3,125 MHz)δ:174.86,150.12,143.88,139.93,138.83,136.92,132.13,131.47,129.52,127.56,124.12,117.82,63.55,29.82,29.27,14.50。
HRMS(ESI): m/z calcd for C20H21N3O2[M+H]+:335.163 4,found: 335.163 8。
化合物6 表征数据:产品为黄色固体,收率77%,熔点123.8℃~125.0℃。
1H NMR(CDCl3,500 MHz)δ:7.46(s,1H),7.45(d,J=8.5 Hz,2H),7.22(d,J=8.0 Hz,2H),7.18(d,J=9.0 Hz,2H),7.15(d,J = 8.5 Hz,2H),6.35(s,1H),4.37-4.26(m,2H),1.39-1.36(s,3H).13C NMR(CDCl3,125 MHz)δ:175.05,144.60,141.95,138.00,134.97,132.03,129.88,127.68,125.81,123.05,118.44,61.56,14.51。
HRMS(ESI): m/z calcd for C18H15Br2N3O2[M+H]+:462.953 1,found: 462.953 5。
1.4 MTT比色法测定化合物的抗细胞增殖活性
将化合物1~6 用DMSO 溶解后稀释,将细胞株SW620(人结肠癌细胞)、MKN45(肺癌细胞)、MKN-28(胃癌细胞)、T47D(人乳腺癌细胞)、3AO(卵巢癌细胞)在96 孔板上种入4 000 个/200 μL/孔,每孔加入化合物2 μL,终浓度分别为100 μM、50 μM、20 μM、10 μM、5 μM、2.5 μM,在细胞培养箱(37℃,5% CO2)中孵育72 h,以DMSO(1%)为空白对照。72 h 后加入终浓度为0.25 mg/mL 的MTT,于细胞培养箱中孵育4 h,之后吸干溶剂,每孔加入100 μL的DMSO,用酶标仪测定570 nm处的吸光度值(OD 值),计算药物对细胞增殖的抑制率以及被测样品的IC50值,公式如下:
抑制率=[1-(测试样品的OD 值-空白OD 值)/(阴性对照的OD值-空白OD值)]*100
lg IC50=Xm-I[P-(3-Pm-Pn)/4],其中,Xm=lg[最大剂量],I=lg[最大剂量/相临计量];P 为阳性反应率之和,Pm为最大阳性反应率,Pn为最小阳性反应率,结果如表3所示。
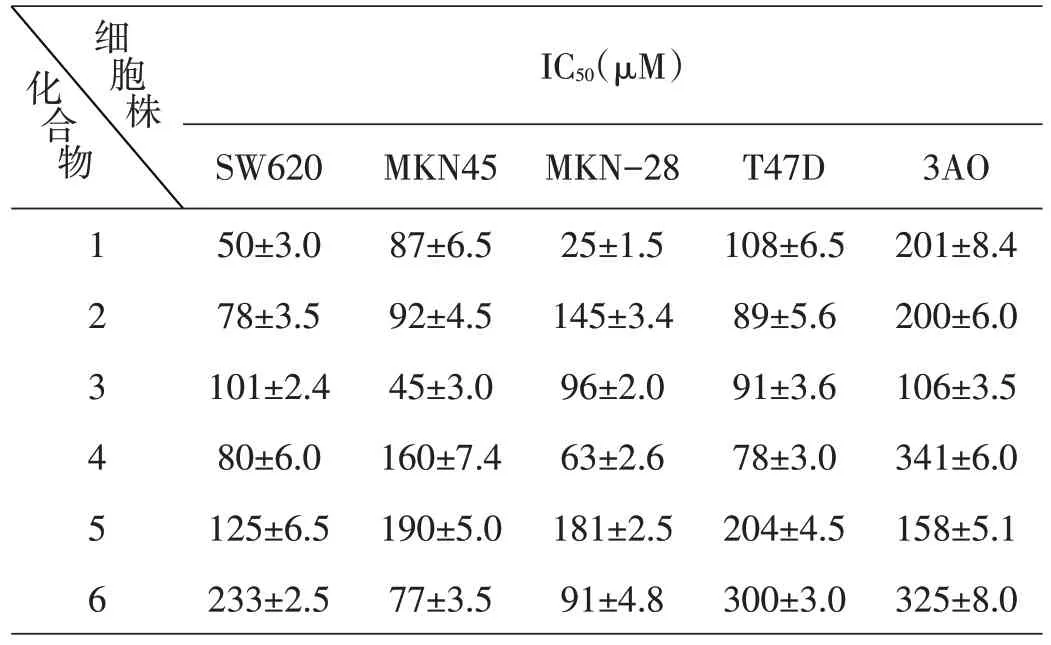
表3 化合物(1~5)对不同肿瘤细胞株的IC50
实验结果表明,该系列化合物对肿瘤细胞活性有较强的抑制功能,特别是化合物1 对SW620(人结肠癌细胞)、MKN45(肺癌细胞)、MKN-28(胃癌细胞)均有明显的效果。
2 结论
本文设计的一锅法制备5-氨基咪唑化合物的合成路线有以下优势:①原料简单,反应条件温和;②实验步骤少,反应操作方便,因为该实验为一锅煮的方法,减少了中间体的分离纯化步骤,降低了成本;③收率高,具有推广应用价值;同时,5-氨基咪唑类化合物在医药领域具有重要的应用价值。本文通过MTT比色法测定了化合物的抗细胞增殖活性,为其在医药方面的应用提供了思路。
