一种具有光致变色性能的有机-无机超分子材料的合成与性能研究
2024-03-05张书泉陈颖程云龙曹若雨
张书泉,陈颖,程云龙,曹若雨
(福州大学至诚学院化学工程系,福建福州 350002)
光致变色材料是一类能在外界光源刺激下发生颜色变化的材料[1-2]。通常来说,这类材料存在两种相对稳定的化学状态(光照前态/光照后态),并且每种形态具有不同的颜色,表现为紫外-可见光谱具备不同的吸收带。在收到一定波长的光照之后,光照前态可以转变为光照后态,并伴随着颜色(吸收光谱)的变化。同时,在受热或与空气中氧气等物质发生作用之后,光照后态又能恢复到原来的光照前态。因此光致变色材料在变色窗户、变色装饰、光开关或光致变色无墨印刷可擦材料等方面具有潜在应用前景。
传统的纯有机光致变色材料如俘精酸酐、偶氮苯、螺吡喃和二芳基乙烯等[3],由于其具有稳定性差、抗疲劳性不强以及对热不稳定等缺点,严重限制了其实际应用[4]。相较于纯有机光致变色材料,近年来受到越来越多关注的有机-无机超分子光致变色材料表现出更优越的性能[5-6]。有机-无机超分子光致变色材料是将具有光致变色的有机组分和能够促进其光致变色的无机组分通过共价键、配位键、氢键、阴离子-π、π-π 相互作用等自组装而成的晶态材料。在这种材料里,光致变色基元由于被保护于密堆积的晶体结构中,因而有效地提高其变色状态的稳定性。这类光致变色材料可以在常温条件和空气中保持稳定的变色状态,在一定温度下加热处理或者暗放置后,能恢复到变色前的状态,重复多次不会出现明显的衰减[7-8]。同时晶态结构的有序性,为研究这类材料的光致变色机理和构效关系提供了可能。
紫精类化合物(Viologens,图1)是一类具有独特氧化还原性能的联吡啶鎓盐。紫精类化合物的联吡啶环表现为缺电子特性,它能在富电子物种的协助下,通过光或热或电刺激,从富电子物种得到电子,形成紫精自由基,这种紫精自由基往往呈现出丰富绚丽的颜色[9]。同时,由于得到的电子在整个紫精π-共轭体系中离域,因此其非常稳定,能在无氧环境长期存在。当在空气或含氧环境中,其自由基被氧捕获,从而被氧化恢复成联吡啶鎓盐,相应的颜色也回到原始状态。金属卤化物由于卤素上具有较多的孤电对,通常被认为是一类优异的富电子物种(电子给体)。因此将紫精类化合物与金属卤化物组装成有机-无机超分子材料,在近年来已经成为开发新型光致变色材料以及研究光致变色机理的重要手段[10]。

图1 紫精类化合物的结构及其自由基的形成过程
本文利用羧基化的紫精衍生物氯化N-(3,5-二羧基苯)-4,4′-联吡啶鎓(H2L·Cl,结构见图2)与无机铋卤化物进行超分子自组装,得到了一例有机无机杂化光致变色材料[(H3L)2(Bi2Cl10)](1)。该材料在光照下可以由黄色转变为绿色,并在空气条件下发生可逆的褪色现象。通过粉末衍射仪、紫外-可见漫反射光谱仪、红外光谱仪、电子顺磁共振波谱仪等方法,对材料的光致变色机理进行了研究。
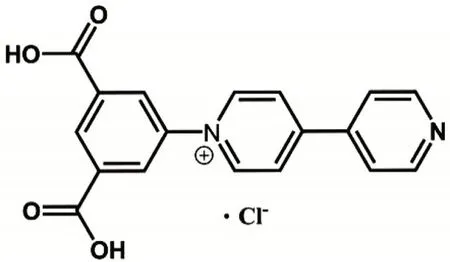
图2 氯化N-(3,5-二羧基苯)-4,4′-联吡啶鎓盐结构式
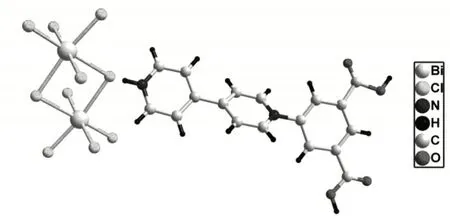
图3 化合物1的晶体结构图
1 实验部分
紫精衍生物H2L·Cl 的制备方法类似于文献所报道[11]。Bi2O3、无水乙醇及盐酸均购自国药试剂,未纯化。
紫精-氯化铋光致变色材料的制备:在10 mL 内衬聚四氟乙烯高压反应釜中加入Bi2O3(0.248 g,0.5 mmol)与H3L·Cl(0.064 g,0.2 mmol)与5 mL蒸馏水,于磁力加热搅拌器上搅拌5 min,随后加入0.6 mL 浓盐酸。继续搅拌30 min后,装入高压反应釜,置于120℃烘箱,恒温3 天。自然降温后,得到无色棒状晶体,过滤并用乙醇清洗3次,于烘箱中烘干,产率为82%。
光致变色及褪色实验:
(1)变色实验:将紫精-氯化铋光致变色材料研磨均匀后,均匀分散在定性滤纸上,并放在培养皿里面。将培养皿放置在氙灯光源下方约2~3 cm处,光照一定时长(对于粉末衍射、红外与顺磁共振分析,样品光照时间为15 min;对于固体紫外光谱分析,光照时长为5 min、15 min、25 min)。
(2)褪色实验:将已经变色的紫精-氯化铋放置在100 mL圆底烧瓶中,插上三通玻璃塞,三通塞一端连接真空泵,一端连接氧气瓶(气路终端阀门)。先用泵抽真空,再通入氧气,反复处理3 次。最后将一个装有氧气的气球绑在三通塞的一端,让烧瓶与氧气球联通,保持这个状态30 min。
图4 为合成的晶体材料的粉末衍射图谱。由图可见,样品的X射线衍射谱图(中间曲线)与从单晶结构拟合出来的谱图(下面曲线)吻合得很好,表明通过水热法得到的晶体为纯相。

图4 化合物1在光照前后的粉末衍射谱图
2 结果与讨论
2.1 紫精-氯化铋光致变色材料的结构表征
紫精-氯化铋光致变色材料的晶体结构通过X射线单晶衍射确定,见图3。其结晶在单斜晶系C2/c空间群,不对称单元内包含一个质子化的紫精配体(H3L2+)以及半个[Bi2Cl10]4-离子簇。
2.2 紫精-氯化铋光致变色材料的光致变色与褪色实验现象
紫精-氯化铋晶体在显微镜下呈现为无色棒状晶体(图5a),纯度较高,反应釜内并没有发现其他固体杂质。将样品粉末(略带微黄的白色粉末)放在300 W 紫外灯下进行光照,发现随着光照时间延长,样品颜色逐渐变成淡黄绿色(图5b)。随后将变色的样品置于氧气环境中进行褪色反应之后,样品颜色又恢复成初始模样(图5c)。

图5 化合物1的(a)晶体形貌;(b)光致变色过程;(c)褪色过程
2.3 紫精-氯化铋光致变色材料的光致变色机理研究
为了研究紫精-氯化铋光致变色材料的光致变色机理,我们通过对材料在光照前后的粉末衍射谱图、红外光谱、固体紫外漫反射光谱与电子顺磁共振光谱进行了对比和分析。
先对比了紫精-氯化铋材料在光照前后的粉末衍射与红外光谱,如图4所示,紫精-氯化铋材料在光照前后的粉末衍射并没有发生明显变化,这样就可以排除掉紫精-氯化铋在光照后晶体结构发生变化的可能性。图6为化合物在光照前后的红外光谱。从图6可以看出,化合物在光照前后,无论是峰位置还是峰数量都没有发生变化,这表明在光照后化合物内部并没有化学键发生变化(键的生成或断裂),这也充分说明光照后紫精类化合物并没有发生结构变化,排除了光化学反应生成了新化合物的可能性。

图6 化合物1在光照前后的红外光谱
其次,我们对化合物在光照前后以及褪色实验后的固体紫外-可见漫反射光谱进行了分析,如图7,在光照前,样品在400 nm以上近乎没有吸收(因此颜色几近白色)。随着光照时间逐渐延长,样品在400~600 nm 范围出现了一个新的吸收带,根据文献报道,这可以归属为紫精自由基的吸收带[12-13]。有趣的是,在褪色实验之后,样品的紫外吸收峰形状又恢复到未光照时的形状。这种现象证实了紫精-氯化铋在光照后通过PIET作用,产生了有色的紫精自由基,从而发生光致变色现象;而紫精自由基被氧气氧化后恢复成紫精分子,从而发生褪色现象。这与前文提到的紫精化合物的光致变色机理相吻合。

图7 化合物1在光照前后以及褪色实验后的固体紫外-可见漫反射光谱
最后,我们对样品在光照前后的电子顺磁共振光谱进行了分析。众所周知,顺磁共振光谱能够有效地对自由基进行检测。如图8所示,在光照前,在3 506.9 G有一个微弱的自由基信号,而在光照后,该自由基信号大大增强了。与文献报道的紫精类化合物进行对比[14-15],该自由基可以归属为接收了一个电子的紫精自由基(H3L·+)。光照前之所以有微弱的自由基信号,可能是在显微镜下观察样品时,照明的白光灯使样品产生了轻微的PIET 作用,从而生成了少量紫精自由基。顺磁共振光谱为光照后通过光诱导电子转移作用产生紫精自由基提供了直接的证据。
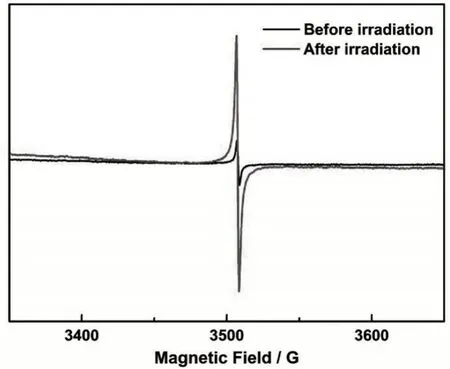
图8 紫精-氯化铋在光照前后的电子顺磁共振光谱
2.4 紫精-氯化铋光致变色材料的变色及褪色机理
对于本实验所制备的紫精-氯化铋光致变色材料的光致变色原理可以解释为:在光照下,电子给体铋氯簇[Bi2Cl10]4-通过光诱导电子转移作用,将电子转移给紫精类化合物(标注为H3L2+),将紫精类化合物还原为自由基H3L·+,同时自身由于失去一个电子,转变为[Bi2Cl10]3-。生成的紫精自由基由于颜色有别于原本的紫精类化合物,因此外观上表现为产品颜色发生变化。生成的紫精自由基可以被氧气氧化,从而发生褪色反应,机理为:氧气与自由基H3L·+反应生成氧负离子O2·-,同时将H3L·+氧化生成H3L2+。而氧负离子O2·-又可以将[Bi2Cl10]·3-转化为[Bi2Cl10]4-,完成一个循环。总的反应机理见图9。
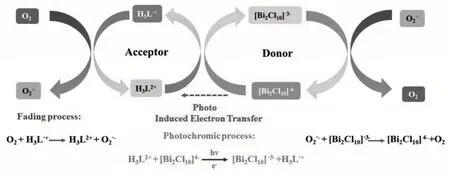
图9 紫精-氯化铋光致变色材料的光致变色及褪色机理图
3 结束语
通过水热合成法制备了一种紫精-氯化铋光致变色材料,并用氙灯为光源,通过光照实验与褪色实验,观察了这类材料的可逆光致变色现象。与此同时,通过样品光照前后的X 射线粉末衍射、红外光谱、固体紫外漫反射光谱以及电子顺磁共振光谱对其光致变色机理进行了研究和讨论。
