法莫替丁合成工艺改进
2024-03-05王亮
王亮
(上海市汇伦医药股份有限公司,上海 201100)
法莫替丁(Famotidine),化学名:3-[[2-[(二氨基亚甲基)-4-噻唑基]甲基]硫代]-N-氨磺酰丙脒,是继西咪替丁和雷尼替丁之后推出的第三代胍基噻唑类的H2受体阻滞药,其疗效确切,不良反应少,具有对H2受体亲和力高的特点,其作用机制与西咪替丁相似。法莫替丁可有效地抑制基础胃酸、夜间胃酸和食物刺激引起的胃酸分泌,亦可抑制组胺和五肽胃泌素等刺激引起的胃酸分泌,其抑制H2受体的强度比西咪替丁强20倍,比雷尼替丁强7.5倍[1]。此外,法莫替丁也可抑制胃蛋白酶的分泌,临床用于胃及十二指肠溃疡、应激性溃疡、急性胃粘膜出血、胃泌素瘤以及反流性食道炎等。与质子泵抑制药奥美拉唑相比,法莫替丁拥有较为低廉的价格、稳定的疗效以及较为庞大的患者群体,预计我国法莫替丁市场销售额今后会进一步增长。因此,研究法莫替丁的合成工艺优化具有一定的经济价值。
1 合成路线
法莫替丁的合成路线和制备方法多有文献报道,主要有以下三种方法:
1.1 合成方法一
以脒基硫脲为起始原料,在碘化钾的催化下与1,3-二氯丙酮缩合环合反应生成2-(4-(氯甲基)噻唑-2-基)胍基盐酸盐,接着与氯丙腈缩合反应生成2-(4-(((2-氰乙基)硫基)甲基)噻唑-2-基)胍,水解反应生成甲基3-(((2-((二氨基甲基)氨基)噻唑-4-基)甲基)硫基)丙亚胺甲酯,最后与硫酰胺亲核取代反应生成化合物法莫替丁(图1)。
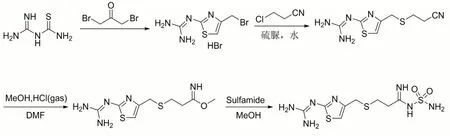
图1 法莫替丁合成路线一
文献[2]报道了该路线,1,3-二溴丙酮价格昂贵,且使用到盐酸气,对设备的腐蚀较大,对环境污染大,得到的法莫替丁原料药成本高昂。
1.2 合成方法二
以3-(甲硫基)丙腈为起始原料,与甲醇、氯化氢反应生成3-(甲硫基)-丙酰亚胺甲酯,接着与硫酰胺反应生成(Z)-3-(甲硫基)-N′-氨磺酰丙脒,在氨气存在下脱去甲硫基生成N-(硫酰胺基)-2-丙烯脒,再与2-(4-巯基甲基)噻唑-2-基)胍反应得到法莫替丁(图2)。
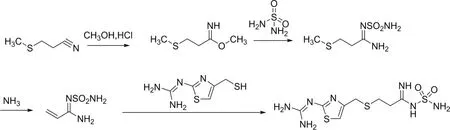
图2 法莫替丁合成路线二
文献[3]报道了该路线,其中(Z)-3-(甲硫基)-N′-氨磺酰丙脒的合成步骤繁琐,后处理复杂,且该中间体不易保存,质量不稳定。该工艺使用到盐酸气,对设备和环境不友好,整体路线收率偏低。
1.3 方法三
文献[4]和文献[5]报道了该路线,此法反应条件温和,转化率高,商品化生产的原料易得,生产周期短,总收率较高。
目前,国内厂家采用的主要工艺路线是方法三,该路线的关键中间体法莫替丁单盐(2-(4-(氯甲基)噻唑-2-基)胍基盐酸盐)的合成工艺报道较少(图3)。本文改进了法莫替丁单盐合成方法,大大提高了法莫替丁单盐的收率和产品质量,并对法莫替丁合成路线的后续其他反应步骤进行了工艺参数筛选,法莫替丁总收率提高至60%以上,合成路线如图4。
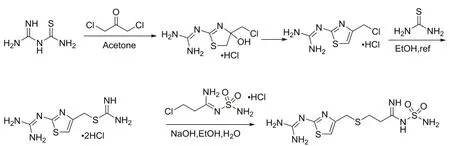
图3 法莫替丁合成路线三

图4 法莫替丁合成路线四
2 实验部分
2.1 仪器和试剂
X-4型显微数字熔点仪;Bruker ARX-400NMR型核磁共振仪,DMSO-d6为溶剂,TMS为内标;四极杆液质联用仪,Agilent 1100;液相色谱仪,1260安捷伦;十八烷基硅烷键合硅胶柱,Symmetry C18 4.6 mm×250 mm,5 μm。
1,3-二氯丙酮,企标,福建仁宏医药化工有限公司;脒基硫脲,企标,山西省芮城县德欣福利香料化工有限公司;硫脲,企标,青州市光大化工有限公司;N-硫酰胺基-3-氯丙脒盐酸盐,企标,无棣融川医药化工科技有限公司;碘化钾,化学纯,山东网化科技有限公司。
2.2 2-(4-(氯甲基)噻唑-2-基)胍基盐酸盐(1)[1]的合成
30 L 反应釜中加入1,3-二氯丙酮(1 000 g,7.94 mol)和5 L丙酮,室温溶解。溶清后,加入10 g碘化钾,降温至-10℃~10℃,分批加入脒基硫脲(936 g,7.94 mol),置于10℃~15℃水浴中搅拌反应4~6 h,过滤,以丙酮(500 mL)洗涤,50℃干燥8 h 得白色固体1 800 g,收率:100%,纯度96.0%。(HPLC 面积归一化法:色谱柱为十八烷基硅烷键合硅胶柱(Symmetry C18 4.6 mm×250 mm,5 μm),流动相为乙腈-甲醇-缓冲盐(1.882 g/L己烷磺酸钠溶液,用乙酸调节pH值至3.5)(94∶6∶900),检测波长为270 nm,柱温25℃,流速1.0 mL·min-1)。mp:163℃~165℃。1H-NMR(400 MHz,DMSO-d6)δ:4.74(s,2H),δ:7.41(s,1H),δ:8.34(s,4H),δ:12.84(s,1H)。ESI-MS m/z:191[M+H]+。
2.3 2-二氨基亚甲氨基-4-脒基硫代甲基噻唑二盐酸盐(2)的合成
30 L 反应釜中加入无水乙醇(12 L),氮气吹扫,开启搅拌,加入1(1 500 g,6.60 mol)和硫脲(600 g,7.89 mol)。开启冷凝器,釜内升温至回流,回流温度75℃~80℃开始计时,回流6 h后取样检测。HPLC检验1反应完全。降温至室温,从反应釜中放料减压抽滤,以无水乙醇(500 mL)洗涤滤饼,50℃真空干燥8 h 得白色固体(1 800 g,90%),纯度99.10%。(HPLC 面积归一化法:色谱柱为十八烷基硅烷键合硅胶柱(Symmetry C18 4.6 mm×250 mm,5 μm),流动相为乙腈-甲醇-缓冲盐(1.882 g/L 己烷磺酸钠溶液,用乙酸调节pH 值至3.5)(94∶6∶900),检测波长为270 nm,柱温25℃,流速1.0 mL·min-1)。1H-NMR(400 MHz,DMSO-d6)δ:4.57(s,2H),δ:7.26(s,1H),δ:8.27(s,4H),δ:9.42(s,3H)。ESIMS m/z:229[M-H]-。
2.4 法莫替丁粗品(3)[2]的合成
30 L 反应釜中加入质量分数为95%乙醇(3 000 mL)及水(3 000 mL),开启搅拌,室温下加入2(1 500 g,5 mol),搅拌溶解后,降温至0℃,加入N-硫酰胺基-3-氯丙脒盐酸盐(1 165 g,5.25 mol),于5℃下搅拌滴加30%氢氧化钠溶液(3 000 mL),于20℃~25℃下搅拌反应12 h,降温至0℃~5℃,过滤,用1 L纯净水淋洗滤饼,干燥得粗品3(1 350 g,80%),纯度99.50%。(HPLC 面积归一化法:色谱柱为十八烷基硅烷键合硅胶柱(Symmetry C18 4.6 mm×250 mm,5 μm),流动相为乙腈-甲醇-缓冲盐(1.882 g/L 己烷磺酸钠溶液,用乙酸调节pH值至3.5)(94∶6∶900),检测波长为270 nm,柱温25℃,流速1.0 mL·min-1)。1H-NMR(400 MHz,DMSO-d6,δH8.24(1H,s),δH7.34(1H,s),δH6.50(3H,s),δH3.62(2H,s),δH2.71-2.69(2H,t,J=15.2,7.2 Hz),δH2.49-2.46(2H,t,J=15.1,7.8 Hz)。ESI-MS m/z:336[M-H]-,m/z:338[M+H]+。
2.5 法莫替丁的提纯
30 L 反应釜中加入质量分数为95%乙醇(3 000 mL)及水(3 000 mL),开启搅拌,室温下加入法莫替丁粗品(1 200 g,3.56 mol)搅拌,升温至回流,溶清后,加入12 g 活性炭,继续搅拌20 min。过滤除去活性炭,滤液转移至预热的20 L 反应釜中,缓慢降温析晶。待温度降至0℃~5℃时,过滤,滤饼用1 L纯净水和1 L乙醇淋洗,50℃真空干燥12 h 得白色固体法莫替丁精品1 040 g,收率95%,纯度99.92%。(HPLC面积归一化法:色谱柱为十八烷基硅烷键合硅胶柱(Symmetry C18 4.6 mm×250 mm,5 μm),流动相为乙腈-甲醇-缓冲盐(1.882 g/L 己烷磺酸钠溶液,用乙酸调节pH 值至3.5)(94∶6∶900),检测波长为270 nm,柱温25℃,流速1.0 mL·min-1)。mp:163℃~165℃。1H-NMR(400 MHz,DMSO-d6,δH8.24(1H,s),δH7.34(1H,s),δH6.50(3H,s),δH3.62(2H,s),δH2.71-2.69(2H,t,J=15.2,7.2 Hz),δH2.49-2.46(2H,t,J=15.1,7.8 Hz)。ESI-MS m/z:336[M-H]-,m/z:338[M+H]+。
2.6 法莫替丁核磁检测图谱(图5)
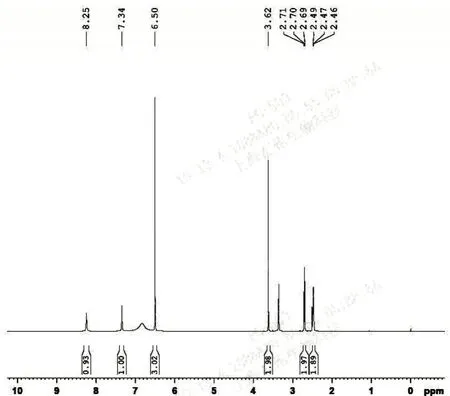
图5 法莫替丁核磁检测图谱
2.7 法莫替丁质谱图谱(图6)
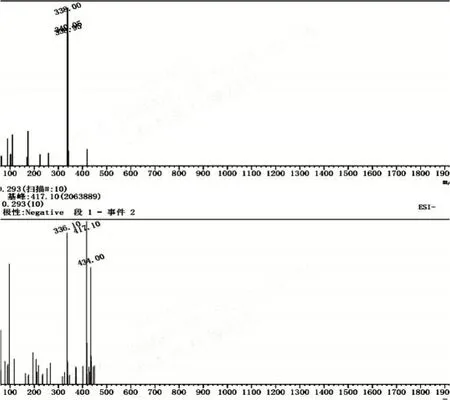
图6 法莫替丁质谱图谱
3 结果和讨论
3.1 法莫替丁中间体1的合成
我们选择非质子溶剂丙酮为反应溶剂,考查了反应温度、反应时间以及反应当量对环合反应的影响。实验结果见表1。
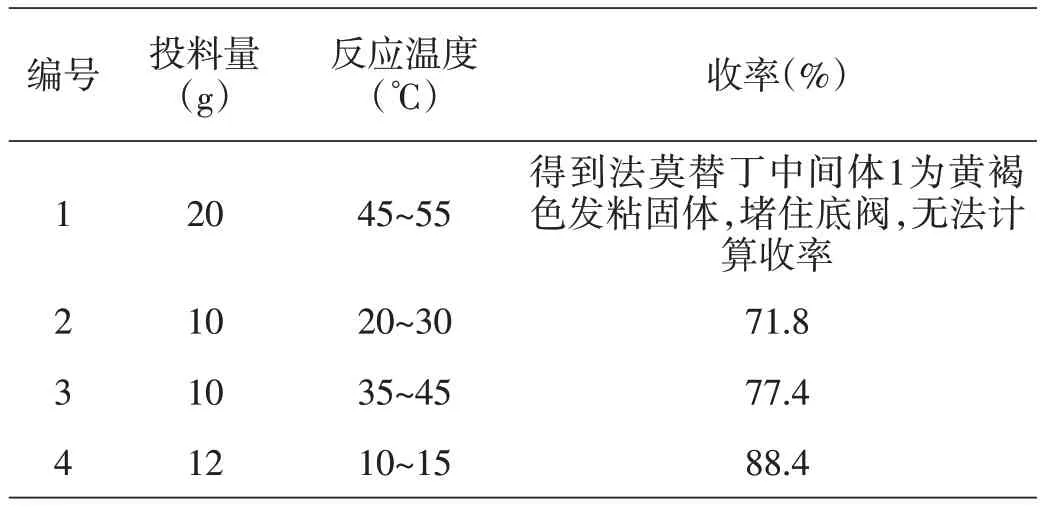
表1 反应温度对合成法莫替丁中间体1的影响
由表1 可知,环合反应温度为45℃~55℃时,产品颜色变深,发粘,无法后续过滤。10℃~15℃下,反应收率高,因此将环合反应温度定为10℃~15℃。产生的副产物经过分离鉴定,主要如图7所示。
将平均粒径18 μm重质碳酸钙分别在900、1 000、1 100、1 200 ℃下煅烧4 h,后采用冶金石灰物理检验方法对石灰活性进行测试,结果如图1所示。

图7 法莫替丁中间体1合成产物及副产物
通过表2 可知,反应6 h 有少量原料未反应完全,产品纯度低;反应8 h 原料反应基本完全,纯度较高;继续延长反应时间至10 h 纯度变低。延长反应时间纯度变低的原因,通过杂质的分离和结构确证,确定是由于部分法莫替丁中间体1 发生水解反应,生成了2-(4-(羟甲基)噻唑-2-基)胍。生成途径如图8所示。
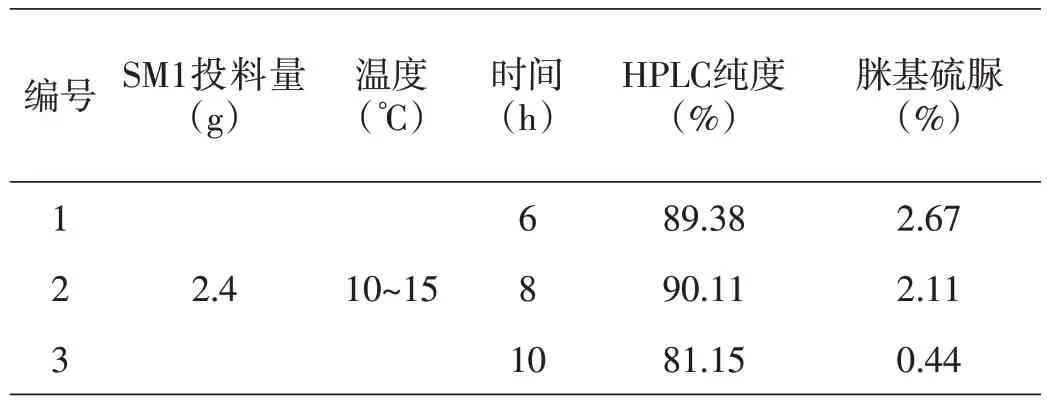
表2 反应时间对合成法莫替丁中间体1的影响

图8 中间体1水解副产物
通过表3工艺筛选数据显示,当SM2与SM1的投料比为0.84∶1 时,132S-1 纯度低,反应收率低;当SM2 与SM1的投料比为0.93∶1时,反应收率较高,纯度也较高;当SM2 与SM1 的投料比为1.03∶1 时,虽然反应收率较高,纯度也较高,但是也增加了SM2的投料量,增加了生产成本。因此选择了SM2与SM1的投料比为0.93∶1。

表3 反应投料比对合成法莫替丁中间体1的影响
综上,环合反应较优的反应条件是将1,3-二氯丙酮和丙酮加入反应釜中,降温至0℃~5℃,分批加入0.93重量的SM1,环合反应温度10℃~15℃,反应6 h,过滤得到法莫替丁中间体1。
3.2 法莫替丁中间体2的合成
该步反应为缩合反应,在合成法莫替丁中间体2的过程中,我们选择无水乙醇为反应溶剂,考查了反应温度、反应时间以及反应当量对缩合反应的影响。实验结果见表4~表6。

表4 反应投料比对合成法莫替丁中间体2的影响
通过表4工艺筛选数据显示,当SM3与132-1的投料摩尔比为1.2∶1时,得到的产品收率和纯度均较好,投料摩尔比1.2∶1为合成法莫替丁中间体2较优的反应投料比。
通过表5 工艺筛选数据显示,反应温度为60℃,得到的产品纯度较低,收率也比回流温度得到的产品收率低,因而60℃为合成法莫替丁中间体2 较优的反应温度。

表6 反应时间对合成法莫替丁中间体2的影响
综上,缩合反应较优的反应条件是将无水乙醇、硫脲和法莫替丁中间体1 加入反应釜中,升温至回流,反应8 h,降温至室温,过滤得到法莫替丁中间体2。
3.3 法莫替丁中间体3的合成
在合成法莫替丁中间体3的过程中,我们选择乙醇和水为反应溶剂,考查了反应温度、反应时间以及反应当量对缩合反应的影响。实验结果见表7~表9。
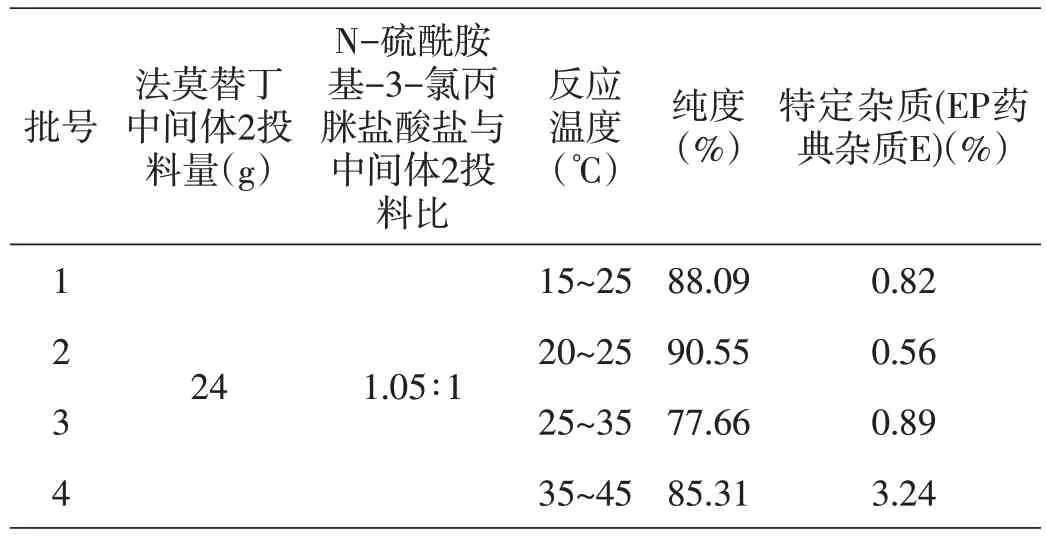
表7 反应温度对合成法莫替丁中间体3的影响
通过表7 工艺筛选数据结果显示,反应温度对反应影响大,物质含量差异明显,35℃~45℃时,杂质E 含量大,后续结晶难以去除;15℃~25℃时,放大生产,有原料反应不完全的风险;综合以上因素,缩合反应温度选择为20℃~25℃。由于反应温度影响的杂质E 在法莫替丁中的残留,且该杂质不易除去,因此该参数列为法莫替丁合成的关键工艺参数。
表8 显示,法莫替丁中间体3 纯度与反应时间差别不大,延长反应时间至6 h后,继续反应,原料变化不明显,因此反应时间4~6 h 为合成法莫替丁中间体3 较优的反应时间。
表9显示,综合纯度和原料中间体2剩余,选择SM4与132S-2的投料比为1.05∶1。

表9 投料比对合成法莫替丁中间体3的合成影响
由以上实验结果得出,中间体3的合成最佳工艺参数:1.05当量的SM4,反应温度为20℃~25℃。
4 结论
(1)法莫替丁中间体1反应温度过低,导致反应周期较长,且主要得到方法三的中间体过高,导致副产物增多,产物转化率变低,产品收率低且纯度不高,对法莫替丁后续合成得到合格的原料药增加了难度。本文采用一锅法制备中间体1,不分离合成路线方法三的中间体2-(4-(氯甲基)-4-羟基-4,5-二氢噻唑-2-基)胍基盐酸盐,直接合成2-(4-(氯甲基)噻唑-2-基)胍基盐酸盐,产品收率达到95%以上,且纯度达到96%以上。
(2)法莫替丁中间体2合成采用质子溶剂乙醇,经检测未检出中间体1与乙醇取代的副产物,该步反应转化率高,副产物少,只要选择合适的反应温度和投料比,即可得到收率和含量都较高的产物。
(3)法莫替丁中间体3 合成,由于N-硫酰胺基-3-氯丙脒盐酸盐在碱性反应体系中极容易自身降解,合适的反应温度可以促进缩合反应的进行和抑制副产物的产生。
(4)法莫替丁中间体3 在乙醇和水的混合溶剂中重结晶,能有效去除杂质,且价格低廉,对环境友好,因此未筛选其他精制溶剂。
综上所述,以脒基硫脲为起始原料,经环合、缩合、缩合精制等四步反应制备法莫替丁,产品纯度为99.92%,总收率为60%,经1H-NMR 和ESI-MS 确证其中间体及目标产物的结构。优化后的合成工艺简单,反应条件温和,产品收率高,纯度高,符合工业化需求。
