脂磷壁酸合成酶ltaS基因缺失对产单核细胞李氏杆菌致病性的影响
2024-03-01胡文洁方小伟田篮鑫
秦 祎,胡文洁,方小伟,郭 骞,田篮鑫,刘 芳,方 春*
(1.长江大学动物科学学院,荆州 434025;2.衡水市农业综合行政执法支队,衡水 053000)
产单核细胞李氏杆菌(Listeriamonocytogenes,LM)是细胞内寄生的革兰阳性菌,也是一种食源性人兽共患病原菌[1]。产单核细胞李氏杆菌具有较强的抗应激能力,例如酸性环境,碱性环境,低温等[2]。产单核细胞李氏杆菌的生存和繁殖所需的养分来自宿主细胞,而如何从宿主细胞中摄取所需的养分,是LM生存的关键因素[3]。产单核细胞李氏杆菌可以通过污染的食物进入胃肠道后扩展至血液循环与淋巴循环进而突破胎盘屏障,血脑屏障等,在其感染过程的每一步均由特定的毒力因子介导(如InlA与InlB)[4-5]。
磷壁酸是革兰阳性菌的一种特殊的化学结构,其中包含了脂磷壁酸(lipoteichoic acid, LTA)和壁磷壁酸(wall teichoic acid, WTA)[6]。脂磷壁酸是革兰阳性细菌中重要的细胞壁聚合物,通常由聚甘油磷酸骨架组成,该骨架通过糖脂与膜相连,使用生物信息学方法已经确定参与糖脂(lmo2555和lmo2554)和LTA骨架(lmo0644和lmo0927)合成的产单核细胞李氏杆菌基因[7]。lmo0927和lmo0644编码的蛋白质与负责聚甘油磷酸骨架合成的葡萄球菌LTA合成酶LtaS具有高度相似性。lmo0644充当LTA引发酶LtaP并将初始甘油磷酸转移到糖脂锚上,而Lmo0927作为LTA合成酶LtaS,延长甘油磷酸骨架[7]。
在产单核细胞李氏杆菌中,磷壁酸能够影响参与穿越宿主屏障的蛋白质与细胞壁共价或非共价相关蛋白。如InlA和InlB都是由细胞表面蛋白或分泌蛋白组成的,它们能够转移到细胞外并且锚定在特定部位,这对产单核细胞李氏杆菌的功能起着关键作用。虽然参与产单核细胞李氏杆菌蛋白转位的系统有六个,如菌毛蛋白装置(FPE)、鞭毛输出装置(FEA)、双精氨酸分泌系统(Tat)以及Sec转位系统等[8],但是Sec分泌系统是最重要的蛋白转位系统之一。InlA和InlB都是通过Sec体系进行转位的,在InlA转位后,其LPXTG基因的基因序列被用 SrtA和细胞壁进行了共价的连接[9]。而InlB经折叠处理后,以非共价态固定于GW基链上的细菌表面,并且包含LTA和WTA。本文构建脂磷壁酸合成酶ltaS基因缺失株,并且通过Western blot试验、细胞黏附侵袭试验、细胞吞噬与胞内增殖试验、小鼠致病性试验来分析脂磷壁酸合成酶ltaS基因的缺失对产单核细胞李氏杆菌InlA和InlB锚定中的作用以及对产单核细胞李氏杆菌毒力的影响。
1 材料与方法
1.1 材料
1.1.1 主要材料 产单核细胞李氏杆菌EGDe-prfA*(血清型为1/2a型),穿梭质粒pKSV7由长江大学动物病原微生物实验室保存。鸡成纤维细胞DF-1细胞和小鼠巨噬细胞RAW264.7细胞由长江大学动物病原微生物学实验室保存。BALB/c小鼠购自长江大学医学院实验动物中心。
1.1.2 主要试剂 脑心营养肉汤(BHI,HB8297-5)从青岛高新技术园区海博生物科技有限公司购入;HRP标记山羊抗兔 IgG及 ECL显影液从上海生工生物科技有限公司购入;核酸染料 Goldview从北京索莱宝科技有限公司购入;Onestep无缝克隆试剂盒从北京康普汇维科技有限公司购入;琼脂糖凝胶回收试剂盒、质粒提取试剂盒及 DNA Marker购自武汉擎科生物科技有限公司。
1.2 方法
1.2.1 缺失株的构建 缺失株构建方法按参照文献进行[10-11],基于产单核细胞李氏杆菌EGDe基因组(登录号NC003210)利用Vector NTI软件设计目的基因ltaS(lmo0927)缺失株所需的引物(表1);首先用引物pFL018-A/B和pFL018-C/D扩增出ltaS上游及下游的同源臂。将第一轮PCR产物共同回收纯化作为 PCR模板,用pFL018-A/D引物进行融合同源臂。纯化融合的同源臂,连接经过BamHⅠ线性化载体pKSV7构建重组穿梭质粒pFL018。使用电转法将重组穿梭质粒pFL018转入亲本株感受态细胞中,经41 ℃的连续氯霉素抗性传代进行同源性重组,再28 ℃连续无抗传代消除质粒,使用旁侧引物pFL018-A/E PCR鉴定缺失株ΔltaS构建成功。
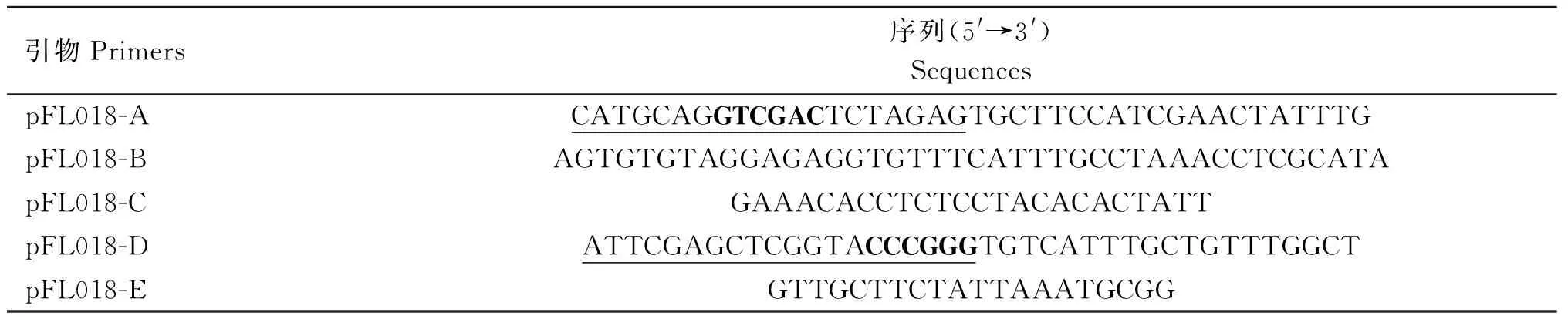
表1 本研究所用的引物
1.2.2 生物学特性的测定 生长曲线试验测定方法按参照文献进行[12],将EGDe-prfA*和ΔltaS挑单菌落于3 mL BHI培养基中,37 ℃摇床220 r·min-1过夜培养。次日取1 mL菌液,12 000 r·min-1离心后用1 mL BHI重悬菌体,使用BHI稀释至10-2,涡旋混匀后取200 μL菌液接于96孔板,每个组设3个平行,于37 ℃恒温培养箱中培养,每间隔1 h使用酶标仪测定OD600 nm值直至细菌生长平台期。
使用革兰染色法观察细菌的形态特征,分别挑取EGDe-prfA*和ΔltaS的单菌落于3 mL BHI液体培养基中,置于37 ℃恒温培养箱过夜培养。次日取1 mL菌液,12 000 r·min-1离心后用1 mL 1×PBS重悬菌体后取10 μL菌液涂布于载玻片中间,干燥、固定后进行染色,初染滴加草酸铰结晶紫水洗后滴加碘液媒染,媒染后使用95%乙醇脱色,最后用番红复染再水洗,自然干燥后使用光学显微镜进行细菌形态的观察。
1.2.3 Western blot检测 蛋白样品制备参照文献进行[13],将菌液划线于无抗BHI平板,置于37 ℃ 培养箱过夜培养;次日挑取单菌落于无抗BHI培养基中37 ℃ 摇床220 r·min-1过夜培养;将培养菌液按1∶50转接于50 mL BHI培养基,置于37 ℃摇床220 r·min-1培养10 h后12 000 r·min-1离心收集上清和菌体沉淀。菌体沉淀用PBS洗涤后加入含2% SDS的PBS重悬,置于37 ℃恒温培养箱静置2 h,间隔10 min涡旋一次,最后12 000 r·min-1离心并收集上清,则为表面蛋白样品。取40 μL蛋白样品加入10 μL的 SDS-PAGE上样缓冲液混匀,煮沸10 min后离心。按相应配方配制12% SDS-PAGE蛋白胶,将预染蛋白 Marker及10 μL样品一次加入胶孔,进行SDS-PAGE分离湿转法转膜(80 min 230 mA)将PAGE胶上蛋白转移至PVDF膜上封闭采用1×TBST配制5%脱脂乳浸泡膜,置于25 ℃的摇床60 r·min-1封闭2 h;用 1×TBST清洗2~3次,然后将膜放入一抗溶液中,一抗为5%脱脂乳(TBST 配制),对应于1∶500的自制InlA、InlB、GAPDH多克隆抗体血清,在4 ℃静置孵育过夜;次日使用 1×TBST清洗膜5次,每次10 min;二抗在5%的脱脂乳与1∶5 000羊抗兔IgG二抗中,置于25 ℃ 60 r·min-1的摇床中培养1 h;二抗孵育完成后,使用 1×TBST清洗5次,每次10 min,将 ECL显影液均匀铺于薄膜表面,并使用化学发光成像系统进行显影。使用软件Image J分析软件对Western blot图像数据进行灰度分析,用内参GAPDH灰度值校正各组分中对应的目的条带灰度值,相对灰度值为校正后缺失株的灰度值与亲本株比值。
1.2.4 细胞黏附与侵袭试验 试验方法按参照文献进行[12],将DF-1细胞以2×105细胞·孔-1接种于24孔板,在5% CO2培养皿中静置过夜,24 h后,用950 μL的无抗生素的细胞培养液替代;添加50 μL的稀释度适宜的菌液,使感染的复数(MOI)大约为10∶1,将其混匀后置于37 ℃ 的5% CO2培养皿内,用平板计数法计数实际感染剂量N0。黏附试验,感染后30 min,弃掉培养液,PBS清洗3次,将1 mL ddH2O加入各个孔,充分吹打使细胞裂解,稀释后用平板计数法计数黏附的菌数目N1。黏附率计算方法为N1/N0×100%。侵袭试验,感染后30 min,弃掉培养液,PBS清洗3次后添加含有50 μg·mL-1庆大霉素的DMEM培养基,置于细胞培养箱中培养1 h后用PBS洗3次,使用相同的方式进行细胞裂解,用平板计数法计数入侵细胞的细菌数量N2,侵袭率计算方法为N2/N0×100%。
1.2.5 细胞吞噬与巨噬细胞内增殖试验 细菌吞噬与巨噬细胞内增殖试验的细胞培养方法与黏附侵袭试验相同。细菌抗吞噬试验,细菌与细胞混匀感染30 min后,弃掉培养液,PBS清洗3次后添加含有50 μg·mL-1庆大霉素的DMEM培养基,置于细胞培养箱中温养1 h,根据细胞黏附侵袭试验弃培养液,清洗,裂解细胞,稀释,用平板计数测定吞噬细胞的数量N3。巨噬细胞内增殖试验,细菌与细胞混匀感染30 min后,弃掉培养液,PBS清洗3次后添加含有50 μg·mL-1庆大霉素的DMEM培养基,置于细胞培养箱中温养4 h,弃培养液、清洗、细胞裂解、稀释,用平板计数法计数细胞的数量N4。吞噬率计算方法为N3/N0×100%;增殖倍数计算方法为N4/N3,试验3次重复。
1.2.6 小鼠致病性试验 将BALB/c小鼠随机分为3组,亲本株组和缺失株组各12只,未感染组5只,饲养一周,使小鼠适应环境。菌液划线于无抗BHI平板,置于37 ℃ 培养箱过夜培养。第二天离心收菌,用PBS缓冲液(磷酸盐缓冲液)洗涤3次,调节OD600 nm至0.6,然后将菌液用PBS稀释100倍。将稀释的菌液经腹腔注射感染小鼠,每只注射0.2 mL(约为4.0×106CFU)用点板计数来测定真实的感染剂量,未感染组的5只小鼠注射0.2 mL PBS,作为对照。在注射后48 h随机选择亲本株组和缺失株组小鼠各7只,在超净台中无菌操作取出小鼠肝和脾,磨碎后在研钵中加入1 mL的PBS缓冲液转移至EP管内,再将组织原液进行梯度稀释点板计算肝和脾细菌数量。各组剩余的5只小鼠,继续观察,记录存活情况,绘制存活曲线。对于LD50试验,将原始菌液(约为2.0×109CFU)梯度稀释至10-2、10-3、10-4三个梯度,每个梯度腹腔接种0.2 mL至BALB/c小鼠,每组8只。使用改良寇氏法计算7 d内亲本株EGDe-prfA*与缺失株ΔltaS对BALB/c小鼠半数致死量LD50。
1.2.7 统计分析 用3个重复试验的平均值和标准偏差来表达结果,用 GraphPad Prism 5作图,采用非配对t检验(unpairedttest)法进行统计学分析,P<0.05表明有统计学差异,差异显著,使用“*”标注;P<0.01表明有统计学差异,差异极显著,使用“**”标注;P>0.05表明差异不显著,使用“ns”标注。
2 结 果
2.1 缺失株的构建
亲本株EGDe-prfA*基因组作为模板,用引物pFL018-A/B和pFL018-C/D,分别获得上游同源臂610 bp、下游同源臂697 bp片段。(图1A);将PCR产物回收纯化后作为模板,用引物pFL018-A/D进行扩增,获得1 307 bp的融合同源臂片段(图1A)。将融合同源臂片段纯化后与线性化的pKSV7重组连接转化后成功构建重组质粒pFL018(图1 B)。使用电转法将重组质粒pFL018转入亲本株感受态细胞中(图1B),经41 ℃的持续性有抗传代来筛选重组后的菌株,并在28 ℃连续无抗传代,以不间断传代的方式消除质粒,使用旁侧引物采用PCR方法鉴定筛选缺失株ΔltaS(图1C)。
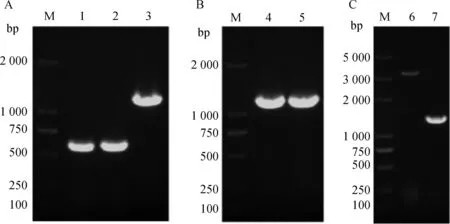
M.DNA 相对分子质量标准;1. 上游同源臂;2. 下游同源臂;3. 融合的同源臂;4. DH5α-pFL018;5.EGDe-prfA*-pFL018;6. 亲本株EGDe-prfA*;7. 缺失株ΔltaSM.DNA marker;1. Upstream homology arm; 2. Downstream homology arm; 3. Fusion homologous arm; 4. DH5α-pFL018; 5. EGDe-prfA*-pFL018; 6. Parent strain EGDe-prfA*; 7. Mutant strain ΔltaS
2.2 ltaS缺失不影响产单核细胞李氏杆菌的生长和菌体形态
体外培养试验结果表明,缺失株ΔltaS与亲本株EGDe-prfA*在细菌迟缓期、对数生长期以及平台期基本一致(图2A)。将缺失株ΔltaS与亲本株EGDe-prfA*进行革兰染色后镜检发现菌体形态无变化(图2B)。
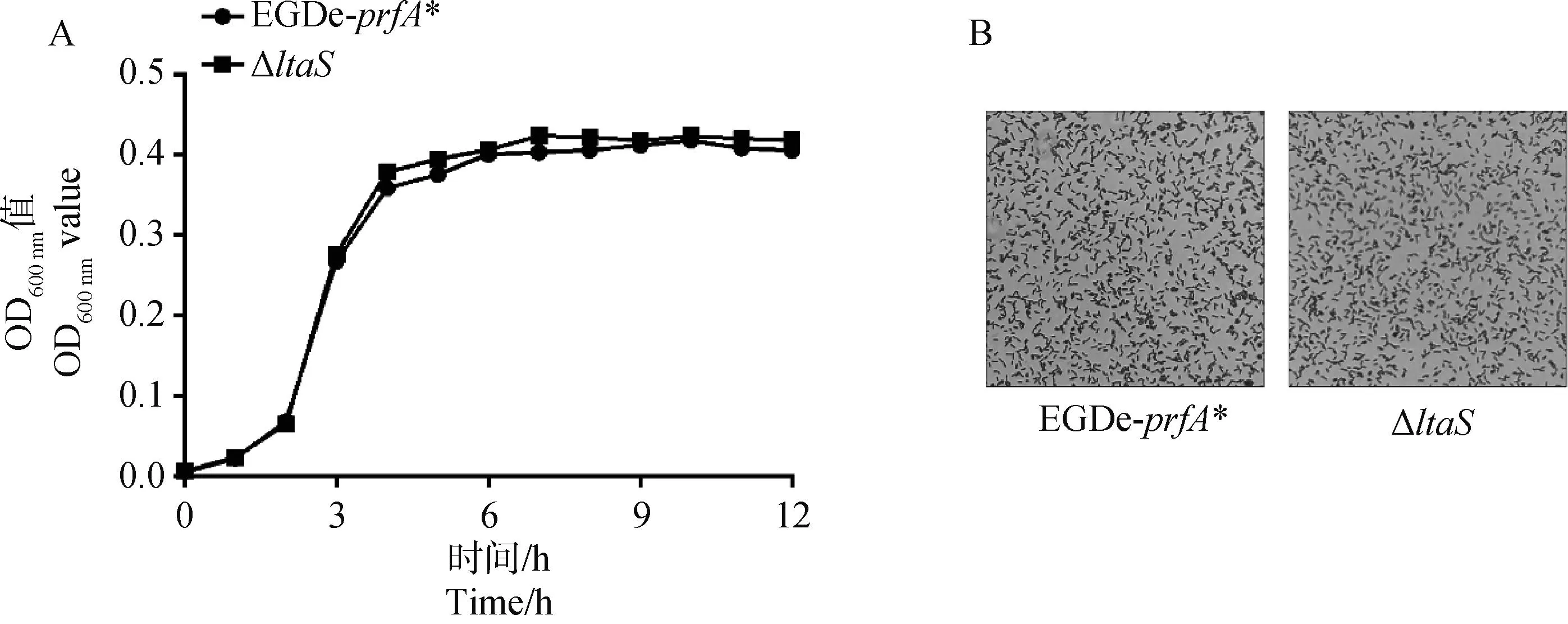
图2 细菌生长曲线测定(A)以及细菌形态观察(B)Fig.2 Determination of bacterial growth curve (A) and observation of bacterial morphology (B)
2.3 ltaS缺失减少产单核细胞李氏杆菌表面锚定的InlB
Western blot试验分析InlA和InlB在产单核细胞李氏杆菌表面的锚定情况,灰度分析结果显示,与亲本株相比,在缺失株ΔltaS表面,InlA和InlB锚定量极显著降低(P<0.01)(图3)。结果说明ltaS基因缺失后显著减少InlA和InlB在产单核细胞李氏杆菌表面的锚定。
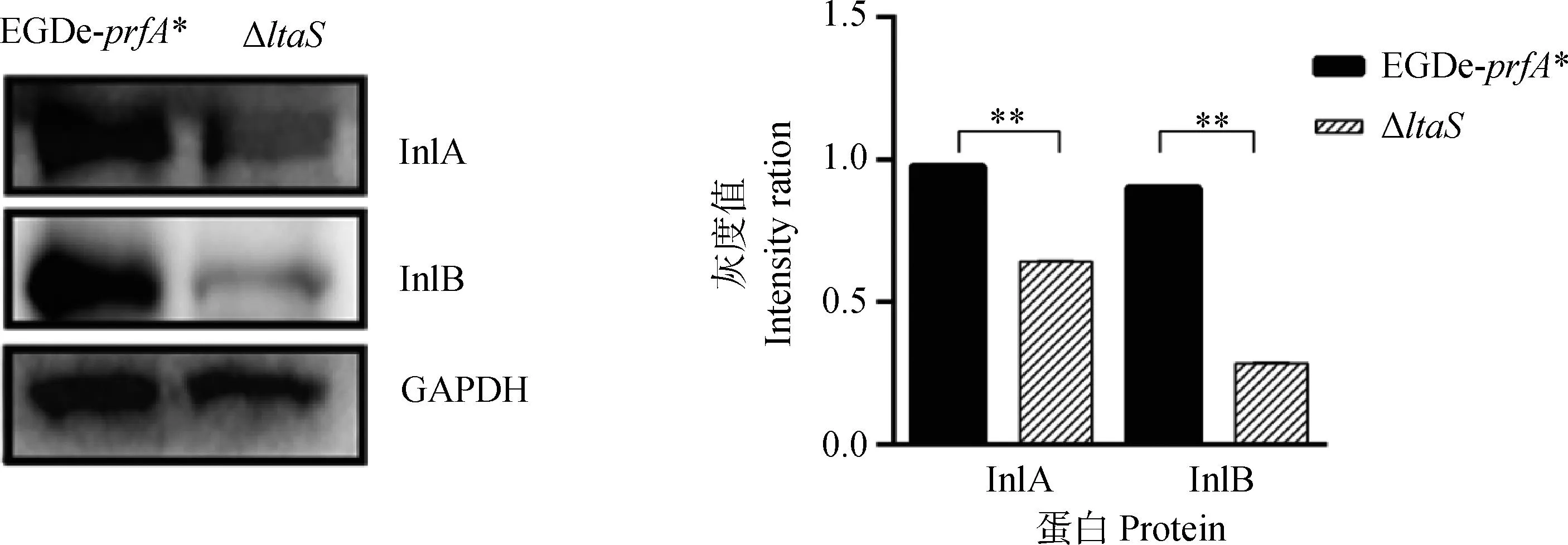
*.差异显著(P<0.05);**.差异极显著(P<0.01);ns.无显著差异(P>0.05)。下同*.Significant difference(P<0.05);**.Extremely significant difference(P<0.01);ns.No significant difference(P>0.05).The same as below
2.4 ltaS缺失显著降低产单核细胞李氏杆菌对细胞的黏附侵袭能力
细胞黏附试验结果表明,缺失株ΔltaS对 DF-1细胞的黏附率极显著低于亲本株EGDe-prfA*的黏附率(P<0.01)(图4A);细胞侵袭试验结果表明,缺失株ΔltaS对 DF-1 细胞的侵袭率极显著低于亲本株EGDe-prfA*的侵袭率(P<0.01)(图4B)。

细菌对DF-1细胞的黏附率(A)和侵袭率(B)Bacterial adhesion rate (A) and invasion rate (B) to DF-1 cells
2.5 ltaS缺失显著降低产单核细胞李氏杆菌在巨噬细胞内增殖能力
试验结果表明,小鼠巨噬细胞RAW 264.7对ΔltaS的吞噬率与亲本株EGDe-prfA*无显著性差异(P>0.05)(图5A);产单核细胞李氏杆菌在巨噬细胞内增殖试验结果表明,在RAW 264.7中,ΔltaS与亲本菌株EGDe-prfA*的增殖倍数存在明显差异(P<0.05)(图5B)。这说明ltaS基因缺失显著降低产单核细胞李氏杆菌在巨噬细胞内的增殖能力。
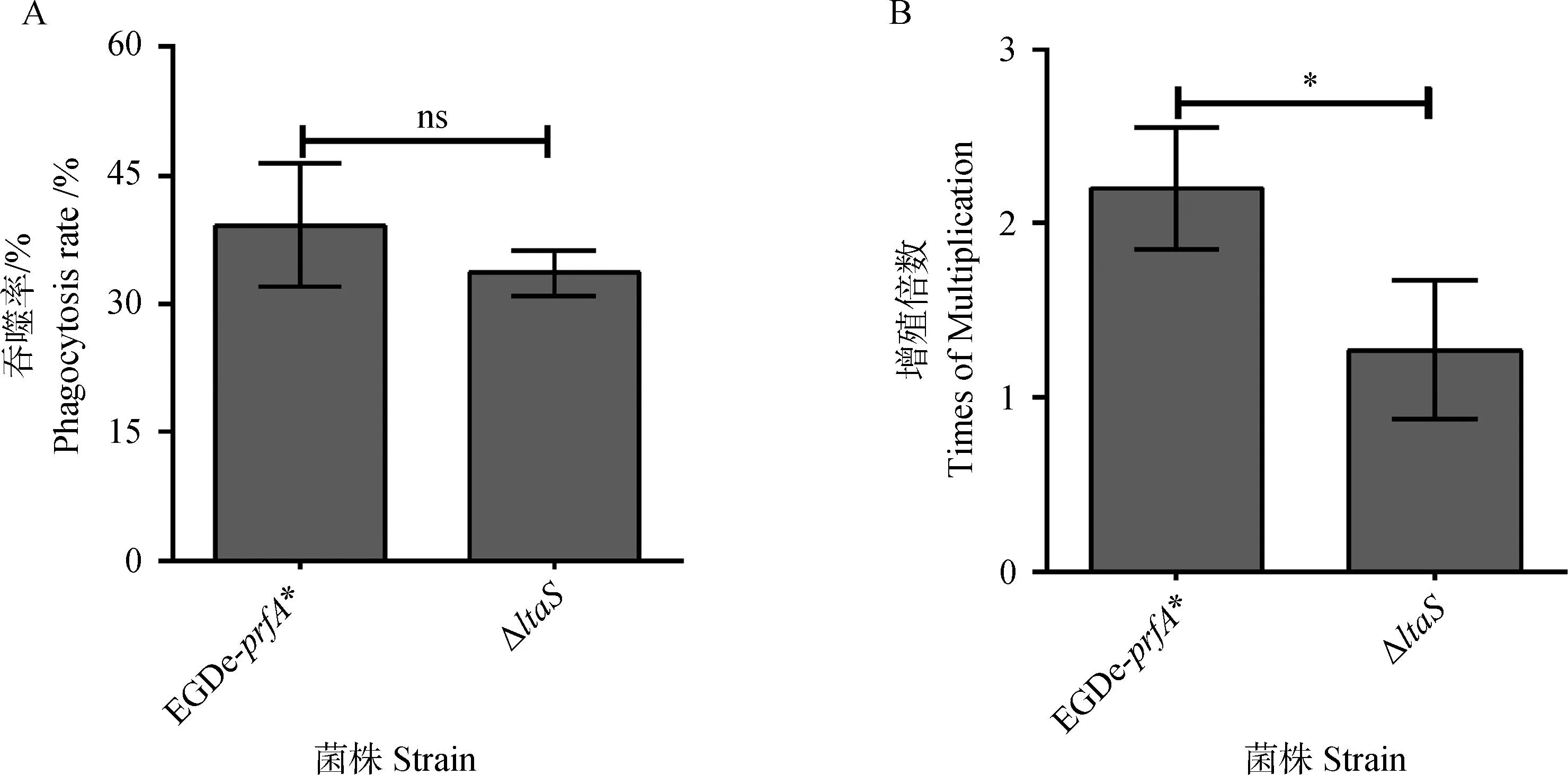
RAW264.7对细菌吞噬率(A)和细菌在RAW264.7细胞中增殖倍数(B)Bacterial phagocytosis rate (A) and bacterial proliferation factor in RAW264.7 cells (B)
2.6 ltaS缺失显著减弱产单核细胞李氏杆菌对小鼠的致病性
以BALB/c小鼠为动物模型去评估产单核细胞李氏杆菌的致病性。小鼠脏器载菌量试验结果表明,感染后48 h内缺失株ΔltaS感染的小鼠肝中的细菌载量与亲本株EGDe-prfA*相比细菌载量极显著降低(P<0.01);与亲本菌株相比,在小鼠脾中的细菌载量也极显著降低(P<0.01)(图6A)。在存活试验中通过改良寇氏法计算得出EGDe-prfA*与ΔltaS对BALB/c小鼠半数致死量LD50分别为1.7×104CFU 和7.49×106CFU。EGDe-prfA*感染后4 d时小鼠全部死亡,ΔltaS感染后4 d还有80%存活率(图6B)。结果表明ltaS基因缺失能降低产单核细胞李氏杆菌致病力。
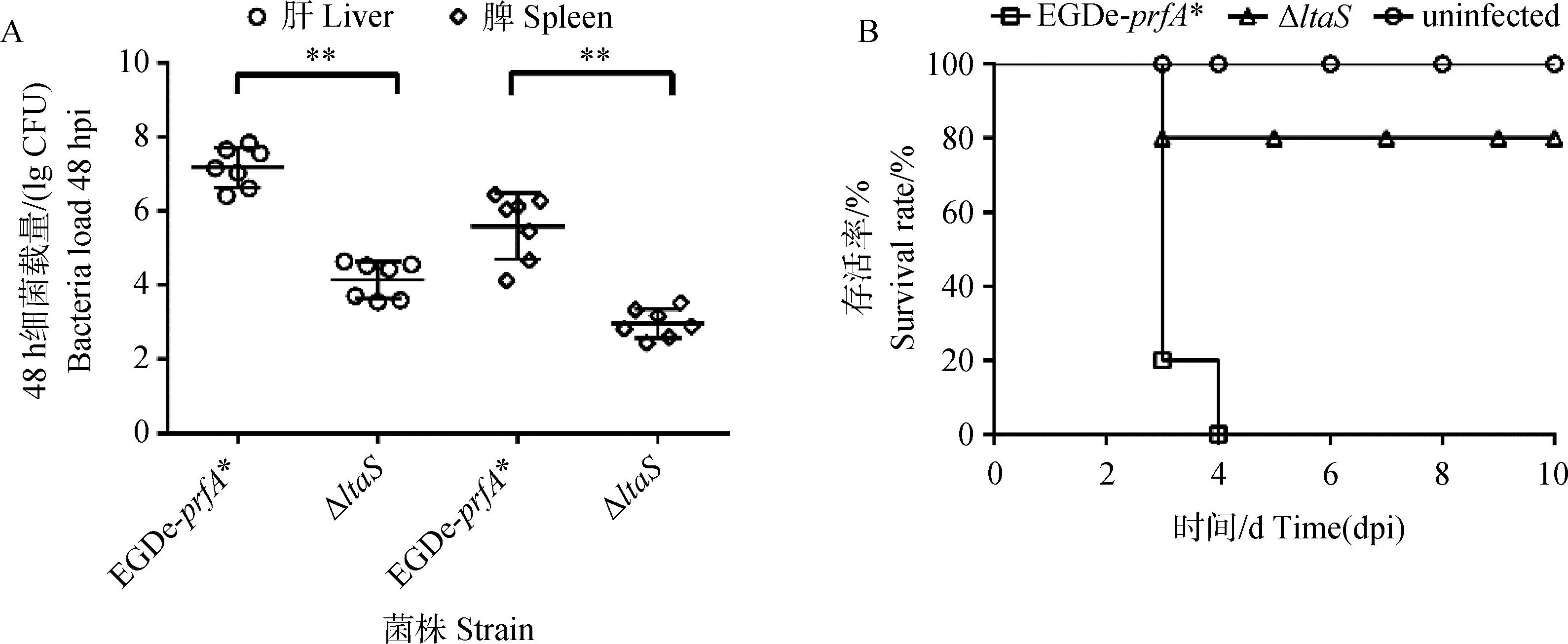
图6 ltaS基因缺失对产单核细胞李氏杆菌感染小鼠肝和脾中细菌载量(A)以及存活率(B)的影响Fig.6 Effects of ltaS gene deletion on bacterial load in liver and spleen (A) and survival rate (B) of Listeria monocytogenes infected mice
3 讨 论
产单核细胞李氏杆菌是革兰阳性食源性致病菌,通过消化道感染人体,并且可以突破宿主三大屏障,胎盘屏障、肠道屏障以及血脑屏障,可引发孕妇流产,宿主脑膜炎、败血症,严重的会导致死亡[14]。LTA是由多个核糖醇连接而成的大分子聚合物,是革兰阳性菌(G+)细胞壁上的一种具有高度免疫能力的特殊多聚体,它是由磷酸氢核糖醇和/或磷酸甘油酯与细胞膜的脂质共价键合而成,为两性分子,一端与细胞膜上的脂质共价键合,而另一端则通过细胞壁向外伸展[15-17]。LTA是革兰阳性菌细胞壁所特有的,研究表明LTA对革兰阳性菌细胞壁的修饰很重要而细胞壁的功能修饰往往会对毒力因子锚定产生影响[18]。在产单核细胞李氏杆菌中脂磷壁酸是由脂磷壁酸合成酶ltaS和脂磷壁酸引物酶ltaP两个基因编码产物合成。
本研究利用同源重组技术成功构建脂磷壁酸合成酶ltaS基因缺失株,评估亲本株EGDe-prfA*与缺失株ΔltaS生长能力与细菌形态,对毒力因子锚定、细胞黏附侵袭率、细胞吞噬能力和巨噬细胞内增殖能力以及对小鼠致病性等生物学特性。生长曲线试验表明ltaS基因缺失并不影响产单核细胞李氏杆菌在BHI中的正常生长,并且光学显微镜下观察细菌形态大小无差别。Western blot试验结果表明ltaS基因缺失会极显著降低产单核细胞李氏杆菌细胞壁表面InlA与InlB蛋白含量。InlA与InlB是在产单核细胞李氏杆菌感染过程中重要的毒力因子,有帮助其穿越宿主胃肠屏障的功能。InlA介导细菌通过E-钙黏蛋白入侵细胞,而InlB则是一种特异性更低的入侵蛋白[19-20]。研究表明,InlB与LTA结合主要是与该蛋白质和该聚合物的聚甘油磷酸骨架的特异性相互作用[21-22],并且两者以非共价锚定方式在细菌表面[23-24]。当ltaS基因缺失时会导致LTA骨架合成失败,进而导致InlB在细胞表面锚定量降低。然而InlA和InlB是由同一操纵子编码的蛋白质,InlB是InlA依赖性侵袭途径中的促进者,InlB锚定量的降低会影响InlA对细胞侵袭量[25-27]。这也可能是InlA与InlB在缺失株ΔltaS中锚定量降低的主要原因。
体外细胞试验结果表明,ltaS基因缺失会极显著影响产单核细胞李氏杆菌对DF-1细胞的黏附侵袭能力,这与前面Western blot结果相符的。由于ltaS基因缺失导致胞外InlA与InlB含量减少,产单核细胞李氏杆菌黏附侵袭能力减弱。ltaS基因缺失不影响巨噬细胞RAW264.7对产单核细胞李氏杆菌的吞噬能力,但是在巨噬细胞内增殖能力极显著减弱。体外动物致病性试验通过小鼠脏器载菌量以及小鼠存活试验表明,ΔltaS感染的小鼠肝与脾中的细菌载量与亲本株EGDe-prfA*相比细菌载量极显著降低。Webb等[7]提出双酶系统进行 LTA 合成,发现产单核细胞李氏杆菌内两个 LtaS 旁系物的不同酶促功能。在没有LtaP的情况下,LtaS可以使用糖脂作为锚点。但是ltaS缺失影响细菌表面LTA合成,在Δlmo0927中完全不存在LTA[7]。DltABCD基因家族主要功能是对LTA进行D-丙氨酰化修饰,通过催化D-丙氨酸残基与细胞壁脂磷壁酸结合。当LTA合成减弱时,Dlt家族基因无法进行修饰导致无法正常发挥功能。Abachin等[28]发现DltABCD失活会导致产单核细胞李氏杆菌在小鼠肝与脾载量降低,并且在巨噬细胞(BMM)内增殖能力减弱,推测可能是由于ltaS基因缺失影响Dlt家族基因功能导致产单核细胞李氏杆菌致病性减弱。
4 结 论
ltaS基因缺失会减少产单核细胞李氏杆菌表面毒力因子InlA和InlB的锚定量并且会减弱对DF-1细胞的黏附侵袭能力。ltaS基因缺失会减弱产单核细胞李氏杆菌在小鼠脏器载量并且降低致死率。本研究将为脂磷壁酸合成酶ltaS基因后续的机制探索奠定基础。
