A型流感病毒诱导细胞炎症反应的研究进展
2024-03-01孙怡朋
高 欣,孙怡朋
(中国农业大学动物医学院,北京 100193)
流感病毒(influenza virus)属于正黏病毒科,是一种含囊膜病毒,可分为A型流感病毒(influenza A virus, IAV)、B型流感病毒、C型流感病毒和D型流感病毒,其中A型流感病毒能够感染包括人类、猪、马、禽等多种物种,具有高度传染性,是临床上最重要的病毒之一[1]。IAV 基因组包含8个负链RNA片段,根据毒株不同可编码11~14种蛋白质,其中血凝素(HA)是负责病毒入侵呼吸道细胞的主要糖蛋白,可附着在宿主细胞表面唾液酸残基,在病毒吸附和穿膜过程中起关键作用[2]。IAV感染始于上呼吸道,病毒穿过呼吸道上皮的黏液层后入侵细胞,并诱导宿主先天免疫应答,同时受感染的气道上皮细胞募集多种免疫效应细胞,包括中性粒细胞、单核细胞和巨噬细胞,导致宿主过度炎症和局部组织损伤[3]。在免疫能力正常的宿主中,病毒传播仅控制在上呼吸道内,导致轻度疾病,当宿主年龄较大、患有呼吸系统疾病或免疫状态低下时,病毒可从上呼吸道扩散至肺组织,表现为上皮细胞死亡、过度炎症和气道通透性增加,导致病毒性肺炎和急性肺损伤[4]。此外,由于流感病毒发生抗原漂移,即不同毒株的片段在同一宿主中互换时,病毒将具有高致病性并引起全球大流行,造成大量死亡[5]。H1N1、H5N1和H7N9亚型流感病毒在诱导宿主炎症反应中占主导地位,更倾向于感染肺组织,引起弥漫性肺泡损伤。
IAV感染引起宿主死亡的主要原因是宿主发生过度炎症反应,因此抗流感不仅要抑制病毒复制,更要靶向相关炎症因子在细胞间传递,已有多项研究证明,在IAV感染后期,通过靶向NLRP3抑制炎症小体激活,导致白细胞介素-1β(interleukin-1β,IL-1β)和IL-18的分泌受到抑制,对IAV动物模型的恢复有积极影响[6]。目前靶向宿主炎症开发抗IAV新疗法已成为研究热点,本文就IAV感染诱导的炎症反应以及相关免疫调节剂进行归纳总结,为未来抗IAV药物的开发提供依据。
1 对IAV感染的细胞反应
模式识别受体(pattern recognition receptor,PRR)识别IAV后,活化干扰素调节因子3(interferon regulatory factor 3, IRF3)、IRF7以及核因子κB (nuclear factor-κB,NF-κB),导致促炎细胞因子和趋化因子的产生,此外,NOD样受体蛋白3(NOD-like receptor protein 3, NLRP3)、半胱氨酸天冬氨酸蛋白酶-1(caspase-1)、IL-1β和IL-18前体表达也显著增加,触发NLRP3炎症小体组装和激活,诱导IL-1β和IL-18活化并分泌(图1)。与炎症反应相关的炎性介质包括趋化因子、细胞因子以及转录因子,在IAV感染中诱导过度炎症反应,导致宿主组织损伤。
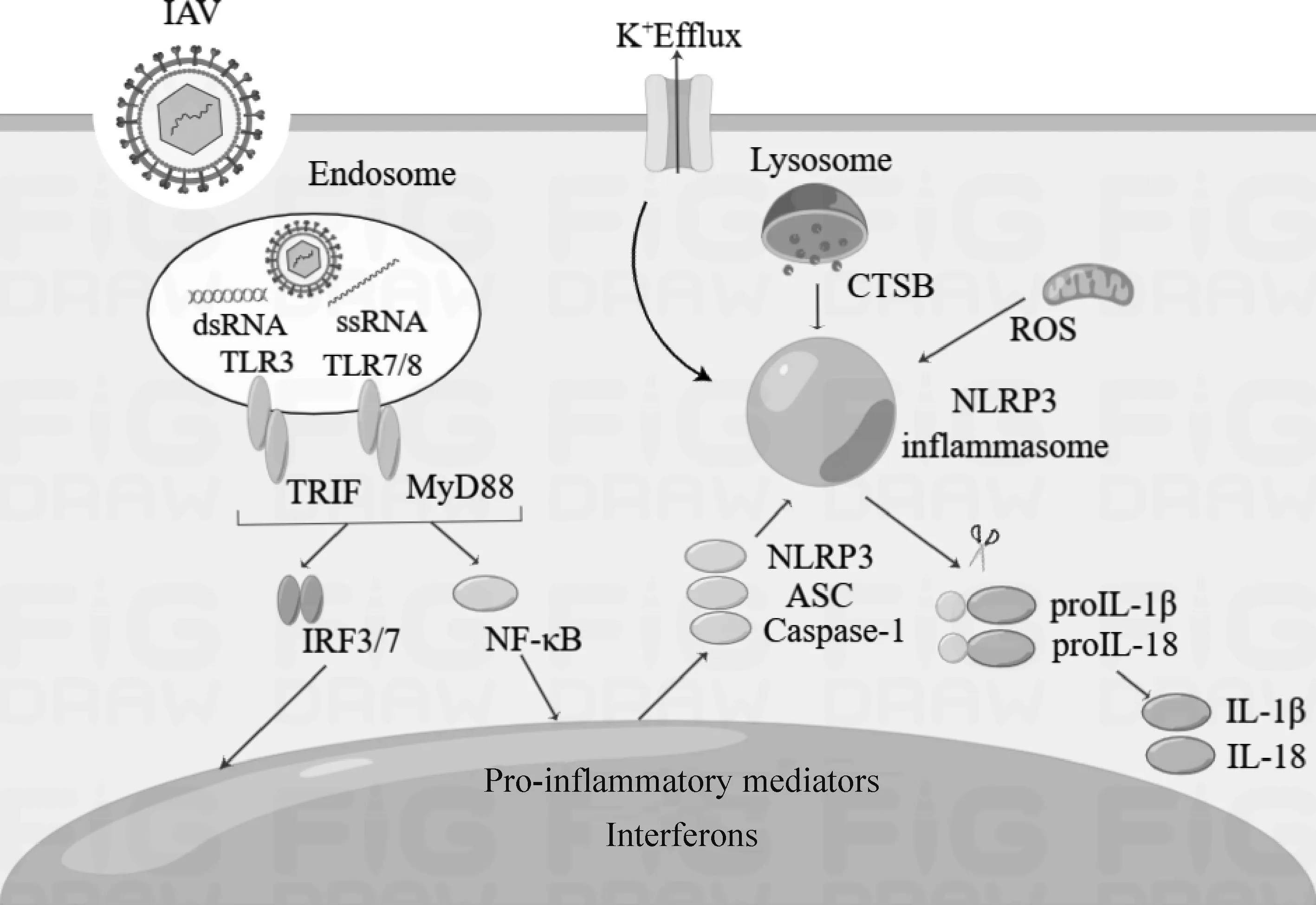
图1 IAV诱导炎症反应示意图Fig.1 Schematic diagram of IAV inducing inflammatory response
1.1 趋化因子
趋化因子是由多种细胞产生的小分子,与白细胞的特定表面受体结合后,改变白细胞形状和行为,从而通过血管内皮迁移到炎症部位,参与维持体内平衡,控制免疫细胞的迁移[7]。在IAV感染期间,免疫细胞的募集对机体免疫反应、病原体清除和组织稳态重建至关重要。在IAV感染早期,趋化因子大量产生,特别是趋化因子C-C基序配体2(chemokine C-C motif ligand 2, CCL2)的表达显著增加,诱导NK细胞和炎性单核细胞的募集,在控制早期病毒复制中发挥重要作用[8]。英国牛津大学Maclean等[9]研究发现,小鼠再次感染IAV之前,常驻记忆B细胞稀疏地散布在小鼠肺组织中,IAV再次感染可诱导趋化因子C-X-C基序配体9(chemokine C-X-C motif ligand 9, CXCL9)和CXCL10的表达,导致表达趋化因子C-X-C基序受体(chemokine C-X-C receptor, CXCR3)的常驻记忆B细胞招募到感染部位,迅速分化为浆细胞,增加局部抗体浓度。然而,趋化因子CCL2的大量产生以及炎性单核细胞的过度募集会增加IAV感染的发病率,同时CCL2受体的抑制会降低小鼠对细菌继发感染的易感性[10]。趋化因子CXCL1和CXCL2也在IAV感染中发挥重要作用,激活后能够募集大量中性粒细胞,使IAV感染诱导的小鼠肺损伤更严重,小鼠死亡率增加[11]。肺上皮细胞产生的CXCL2与IAV急性肺损伤密切相关,是IAV诱导急性肺损伤的主要因子[12]。此外,在IAV和细菌合并感染模型中,CXCL2的过度产生与小鼠肺损伤增加、体重减轻和死亡有关[11]。
1.2 细胞因子
IAV诱发肺炎的一个主要表现即为体内炎症因子过度表达而出现的“细胞因子风暴”[13]。TNF-α被认为是IAV细胞因子风暴中心的促炎细胞因子,负责在IAV入侵后上调IL-1和IL-6的产生,TNF-α升高表明IAV的高致病性,动物试验结果显示,与野生型小鼠相比,TNF-α受体缺失鼠感染H5N1流感病毒后发病率显著下降,但病毒滴度没有显著差异[14]。因此,促炎细胞因子TNF-α在IAV感染中引起机体炎性损伤,靶向TNF-α可能是治疗IAV感染患者的潜在方法。IL-1α和IL-1β是NLRP3炎症小体激活后切割IL-1的产物,通过募集白细胞和诱导其他细胞因子的产生,导致T细胞活化,与IAV引起的肺损伤相关[15]。IL-6是参与机体炎性反应的重要促炎细胞因子,在IAV感染中发挥重要作用[16]。IL-6能够活化内皮细胞,诱导P-选择素、E-选择素和整合素的表达增加,是白细胞募集到肺部所必需的,阻断小鼠IL-6可降低IAV感染小鼠的严重程度[17]。IL-17在IAV感染中也发挥非常重要的作用,在感染早期促进中性粒细胞进入肺部,同时在H1N1感染小鼠募集B细胞[18]。另有一项研究表明,IL-17不能增加病毒清除率,而是通过增强炎症反应促进IAV感染的病理变化[19]。与此一致的是,在H1N1 IAV大流行期间,患者血清中发现高水平的IL-17,导致机体发生严重的炎症反应,发病率显著增加,预后不良[20]。
1.3 转录因子
病毒感染引发炎症反应取决于几种转录因子的激活,包括NF-κB、干扰素调节因子(interferon regulatory factor,IRF)、激活蛋白(activitor protein 1,AP-1)、信号转导及转录活化因子(signal tronsducer and activitor of transcription,STAT)和CCAAT增强子结合蛋白β(CCAAT/enhancer-binding protein β,C/EBPβ)等。NF-κB在炎症反应中发挥重要作用,IAV入侵细胞可导致IKBα/NF-κB解离,从而释放NF-κB并易位到细胞核,调节TNF-α、IL-6等促炎介质的转录[21]。研究发现H5N1 IAV感染患者诱导大量促炎介质均依赖NF-κB[22]。Bernasconi等[23]发现IAV在A549细胞中活化IKK,导致NF-κB的持续激活,产生高水平的IL-8,IL-8诱导炎症和组织损伤。AP-1是MAPK通路的主要底物,主要由c-Fos和c-Jun组成,调节促炎细胞因子的水平,Wan等[24]发现使用AP-1信号抑制剂U0126或c-Jun siRNA处理巨噬细胞可降低IL-1β和caspase-1前体的mRNA,并抑制H3N2 IAV感染时NLRP3炎症小体的活化。此外,不同病毒激活转录因子的方式存在很大差异,这导致不同病毒产生不同的趋化因子和细胞因子。
2 IAV感染诱导炎症反应的机制
IAV基因组编码的几种蛋白中,已报道HA、聚合酶碱性蛋白2(polymerase basic protein 2,PB2)、PB1、核蛋白(nucleoprotein,NP)、基质蛋白2(matrix protein 2,M2)、非结构蛋白1(nonstructural protein 1,NS1)以及PB1-F2在宿主炎症反应中发挥重要作用(表1)[25-31]。

表1 IAV蛋白质诱导的炎症反应
2.1 HA蛋白
IAV 片段4编码HA蛋白,是构成IAV囊膜纤突的主要成分之一,在调节宿主炎症反应中发挥重要作用。Zhao等[25]发现H1N1 HA处理HUVECs细胞后,细胞ICAM-1和IL-6 mRNA和蛋白水平均增加,促进细胞的炎症反应,体内尾静脉注射给小鼠,小鼠肺组织出现明显的弥漫性病理损伤,表现为肺泡间隙扩大伴有肺泡血管扩张,肺泡腔和间质炎性浸润,肺泡腔内红细胞渗出。Nieto等[32]进一步发现在A/California/04/09 (CAL)引入HA S110 L突变在人和小鼠细胞中不会改变IAV pH稳定性、迁移率和入侵细胞能力,对病毒复制能力的影响也较小,但与野毒株相比,HA S110 L突变会显著抑制病毒的致病性,降低小鼠的肺损伤和炎症反应,同时具有更少的炎性细胞浸润。此外,HA含有二硫键,在内质网经过折叠和糖基化形成最终构象。Chamberlain等[33]发现抑制肺上皮细胞中蛋白质二硫键异构酶的活性,降低二硫键的形成,可减少H1N1和H3N2流感病毒HA的寡聚化,显著降低肺上皮细胞的炎症反应,在体内使小鼠缺失蛋白质二硫键异构酶,也可导致IAV感染后炎症反应显著降低。因此,二硫键在HA的功能中发挥重要作用。
2.2 聚合酶
IAV聚合酶是由PB2、PB1和聚合酶酸性蛋白(polymerase acid protein, PA)组成,均已报道参与宿主炎症反应,其中PB2是IAV宿主范围和毒力的主要决定因素。Forero等[26]研究发现,携带1918年大流行IAV PB2的重组病毒感染小鼠可增加病毒的适应性导致小鼠致死率上升,并证明1918年大流行IAV PB2能够增强宿主的炎症反应,促进肺部炎性细胞浸润,抑制对肺再生和修复至关重要的Wnt信号通路,从而促进IAV相关的病理损伤。禽IAV在哺乳动物适应性增强的标志PB2 E627K突变,增强病毒在哺乳动物细胞中复制的同时,也影响了宿主的炎性反应。Yu等[34]发现H9N2禽流感病毒的PB2 627位点发生点突变时在BALB/c小鼠中表现出不同的致病性,这种病理变化与炎症激活因子NLRP3相关,与627K相比,627E的IAV感染后第3天,小鼠肺中NLRP3、IL-1β和TNF-α表达显著升高。DesRochers等[35]发现北美高毒力H7N3和低毒力H7N9禽流感病毒在DBA/2小鼠体内发病机制的差异主要取决于PB2和PA聚合酶基因,H7N3 PB2 E358V突变以及PA P190 S和Q400P突变降低了病毒毒力,同时PB2 E358V突变和PA P190 S突变也导致H7N3禽流感病毒感染后宿主炎症反应降低,小鼠肺组织CCL2、CCL3和IL-1β显著减少。PB1可通过传感器参与宿主炎症反应。Kuriakose等[27]发现双链DNA传感器ZBP1可识别PB1,通过RIPK1-RIPK3-caspase-8触发细胞炎症反应,此外Z-DNA结合蛋白(Z-DNA binding protein 1, ZBP1)能够调节NLRP3炎症小体活化以及促进IAV感染诱导的细胞凋亡、坏死性凋亡和焦亡,ZBP1缺失可降低小鼠在IAV感染期间的炎症反应和上皮损伤,从而免于死亡。
2.3 NP蛋白
IAV病毒蛋白和宿主细胞蛋白之间的相互作用可影响宿主的炎症反应。Kim等[28]发现IAV NP蛋白通过与toll样受体2(toll-like receptor 2,TLR2)和TLR4结合,以剂量依赖性方式诱导BMDM细胞中NF-κB和MAPK的激活,促进促炎细胞因子和趋化因子产生,此外NP还触发caspase-1活化以及NLRP3寡聚化,加剧宿主病理损伤。因此,NP主要通过与宿主蛋白相互作用参与宿主炎症反应。
2.4 M2蛋白
IAV RNA第7段编码M蛋白为基质蛋白,包括M1和M2,其中M2蛋白与NLRP3炎症小体密切相关。NLRP3炎症小体的激活需要两个信号,信号1由TLR或RIG-Ⅰ样受体(retinoic-acid inducible gene Ⅰ-like receptor, RLR)启动,导致caspase-1前体以及IL-1β和IL-18前体上调,信号2是感染或组织损伤产生的应激信号,能够通过离子通道、溶酶体破裂和活性氧(reactive oxygen species, ROS)三种途径促进caspase-1募集NLRP3以及caspase-1、IL-1β和IL-18的成熟,最终激活NLRP3炎症小体[36]。IAV诱导宿主炎症反应的重要机制之一是激活NLRP3炎症小体,信号1为巨噬细胞和树突状细胞中的TLR7信号激活,导致IL-1β和IL-18等促炎细胞因子的合成,信号2为M2蛋白作为离子通道促进K+与Na+外排,导致离子失衡和ROS大量产生,从而激活NLRP3炎症小体[29]。此外,在IAV野毒感染BMDM细胞时,细胞能够分泌IL-1β和IL-18,但当M2蛋白突变失去功能时,IL-1β和IL-18不能正常产生,而将M2重组蛋白刺激BMDM细胞或BMDC细胞可激活NLRP3炎症小体,能够正常产生IL-1β和IL-18以及其他炎性因子[29]。因此,M2蛋白作为离子通道是NLRP3炎症小体活化的重要步骤,在IAV诱导宿主炎症反应中发挥重要作用。
2.5 NS1蛋白
IAV RNA第8段编码NS蛋白,包括NS1和NS2,已有研究表明NS1参与宿主炎症反应过程。Gaba等[30]发现A/Puerto Rico/8/34 (PR8)NS1蛋白能够与混合谱系激酶结构域样蛋白(mixed-lineage kinase domain-like protein, MLKL)相互作用,促进MLKL寡聚化和膜易位,增强MLKL介导的NLRP3炎症小体活化,使IL-β分泌增加,同时MLKL膜易位导致膜破裂和细胞离子稳态丧失,诱发细胞坏死性凋亡。然而,Tao等[37]发现犬H3N2 NS1蛋白通过抑制NF-κB的激活减少促炎细胞因子产生,同时NS1能够与NLRP3直接相互作用,阻断凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing CARD, ASC)寡聚化,从而使NLRP3炎症小体失活,此外,NS1阻断caspase-1前体裂解,使IL-1β和IL-18前体不能成熟,导致宿主炎症反应大大降低。两种不同的研究结果可能与NS1序列相关。Clark等[38]发现2009年大流行的H1N1流感病毒有6个氨基酸发生突变(E55K、L90I、I123V、E125 D、K131E和N205 S),含有这6个突变的NS1蛋白与未发生突变的NS1相比,能够诱导更低水平的宿主炎症反应,减少宿主病理损伤,但不影响细胞或小鼠肺中的病毒滴度。NS1诱导较低水平的炎症可能代表IAV对宿主的适应性增加,为IAV大流行提供了一种新的机制。
2.6 PB1-F2蛋白
PB1-F2由IAV基因片段2编码的非结构蛋白,与病毒的致病性密切相关。Pinar等[31]发现H7N9和A/Puerto Rico/H1N1(PR8)PB1-F2能够显著促进线粒体活性氧的产生,激活NLRP3炎症小体并诱导NLRP3依赖性的肺部炎症和炎性细胞募集,使用NLRP3特异性抑制剂MCC950治疗细胞或小鼠则可显着降低IL-1β成熟,减少炎性细胞募集和细胞因子产生。PB1-F2还可以与宿主蛋白相互作用从而影响IAV的致病性。Leymarie等[39]通过酵母双杂交方法鉴定发现PB1-F2能够与钙结合和卷曲螺旋结构2(calcium-binding and coiled-coil domain 2,CALCOCO2,也称NDP52)互作,进而与TRAF6形成三聚体,增强MAVS诱导的NF-κB活性,促进促炎细胞因子TNF-α和IL-8的表达,引起宿主强烈的炎症反应。PB1-F2影响宿主炎症反应可能与其特殊的结构相关,PB1-F2没有固定结构,在水溶液中形成无规卷曲,在极性溶液中形成富含α螺旋或β折叠的结构,根据细胞类型不同,PB2-F2也存在二聚体、三聚体等多聚体结构,并具有形成淀粉样纤维结构的倾向[40]。Chevalier等[41]通过傅里叶变换红外光谱技术和深紫外测量显微镜确定了IAV感染小鼠中存在PB1-F2淀粉样纤维结构,并给予NF-κB荧光素酶转基因小鼠鼻内滴注单体、淀粉样纤维以及截短形式的PB1-F2,结果发现PB1-F2单体和淀粉样纤维结构能够诱导强烈的炎症反应。因此,PB1-F2诱导的炎性反应与蛋白质序列多态性和寡聚状态密切相关。
3 免疫调节治疗
炎性反应的过度激活与严重的肺损伤和死亡相关,因此可采用免疫调节治疗改善IAV感染引发的过度炎性反应[42]。给与IAV重症患者抗炎药物可能会减轻高水平促炎介质带来的临床症状,减少与IAV感染相关的并发症,并且可以与抗病毒药物协同发挥作用[43]。IAV感染的免疫调节治疗需要遵循几点原则,包括在病毒感染出现症状后使用药物具有显著疗效,针对IAV的抗炎疗法应能够与目前的抗病毒药物协同作用,抗炎疗法应优先降低IAV感染后继发细菌感染的风险,以及抗炎疗法不应干扰宿主抗病毒的能力[44]。表2总结了IAV感染的一些免疫调节策略[45-61]。

表2 IAV感染的免疫调节策略
临床研究中已发现在IAV感染中有效的免疫调节剂。他汀类药物是羟甲基戊二酰辅酶A还原酶抑制剂,属于降脂药物,在临床上广泛应用,近几年的研究发现,他汀类药物能够抑制中性粒细胞募集和促炎细胞因子的分泌从而抑制炎性反应,是在IAV感染中常用的免疫调节药物[62]。研究表明,2009年H1N1流感病毒感染住院患者中给与他汀类药物可降低死亡率[45]。另有研究发现,在小鼠感染前给与他汀类药物可改善肺损伤并抑制H5N1、H3N2和H1N1流感病毒的复制[63]。此外,辛伐他汀可以通过抑制与细胞骨架功能相关的蛋白质来降低促炎细胞因子的水平并减少病毒复制[64]。
另一类在IAV感染期间改善炎性反应的是环氧合酶-2(COX-2)抑制剂,COX-2抑制剂主要作用是镇痛、抗炎和退热[65]。H5N1流感病毒感染可导致上皮细胞COX-2显著上调,抑制COX-2可显著改善小鼠临床症状,与神经氨酸酶抑制剂联合使用效果更佳[46]。在H1N1和H3N2流感病毒感染期间,使用扑热息痛或塞来昔布抑制COX-2可抑制小鼠肺部的病理变化,但不影响病毒的清除率[66]。然而,在人类患者中缺乏此类药物的临床数据,因此COX-2抑制剂的作用有待进一步验证。
在实验性IAV感染的背景下,过氧化物酶体增殖物激活受体(proxisome proliferator-activated receptor,PPAR)激动剂也具有抗炎作用。PPAR激动剂是代谢紊乱(如糖尿病)的治疗药物,PPAR激活后可抑制NF-κB、AP1和STAT信号通路,导致促炎细胞因子减少[67]。H2N2流感病毒感染小鼠模型中,PPAR激动剂吉非罗齐可发挥抗炎作用,增加小鼠的存活率[68]。吡格列酮和罗格列酮也是治疗IAV感染的免疫调节药物,能够减少季节性H1N1流感病毒感染模型中的小鼠体重和致死率。此外,曲格列酮显著降低了IAV感染诱导的肺部损伤[47]。促消退介质也是控制IAV感染诱导炎性的潜在药物。其中脂氧素和保护素是脂质介质,具有抗炎和促消退的作用,在IAV感染中具有保护作用[69]。脂氧素相关基因下调将导致促炎细胞因子上调,促进病毒传播到其他器官。保护素作为炎性反应的调节剂,可改善感染H5N1流感病毒小鼠的存活率,抑制IAV复制[48]。已有研究表明,阻断血小板活化因子受体(platelet activating facor receptor, PAFR)降低小鼠炎性反应,提高感染小鼠的存活率,同时在感染后3 d使用PAFR拮抗剂,仍然具有抑制小鼠炎性反应的作用[49]。此外,PAFR拮抗剂和奥司他韦联合使用能够显著降低感染小鼠的致死率。
细胞受体在炎性反应中发挥重要作用。CC趋化因子受体2(CC chemokine receptor 2, CCR2)是多种白细胞的趋化因子受体,能够募集单核细胞,从而加剧IAV感染期间宿主的炎性反应,引起肺损伤[70]。CCR2的抑制剂可降低IAV感染诱导的病理、发病率和死亡率,而不影响病毒清除率[50]。可作为IAV感染治疗靶点的另一个受体为鞘氨醇-1-磷酸受体(S1P),S1P是鞘脂的代谢产物,与免疫应答相关,参与多种生物过程[71]。使用S1P配体可显著降低细胞因子和趋化因子水平,改善感染小鼠的组织损伤,但不影响中和抗体的产生,也不会改变病毒清除率[51]。此外抑制补体活化、大环内酯类、PDE4抑制剂、罗布麻宁、过氧化氢酶、N-乙酰基-L-半胱氨酸、A1-AdoR、信号通路抑制剂均具有免疫调节作用。综上所述,调节炎性反应是改善重症IAV的潜在策略。
4 展 望
综上所述,IAV感染可诱导宿主气道高促炎性免疫反应,上调IL-1β、IL-6等炎症因子水平,可导致多器官病变,严重者甚至死亡。IAV参与宿主炎性反应与其编码的蛋白质相关,但多集中于表型研究,对机制的研究较少。目前已发现多种免疫调节剂可抑制宿主炎症反应,可能对IAV治疗有效,但临床研究样本量不足,局限于动物试验或体外试验,且不能完全有效。因此,阐明IAV感染涉及的具体炎症机制对开发和改进更有针对性的免疫调节剂至关重要。
