藏药阿如久巴丸质量标准研究
2024-02-23次仁旺姆杨丽琼达娃卓玛
谢 平 次仁旺姆 杨丽琼 达娃卓玛
西藏自治区食品药品检验研究院/国家药品监督管理局中药(藏药)质量控制重点实验室,西藏 拉萨 850000
阿如久巴丸(散)系藏族验方,目前生产厂家有金哈达藏药厂、西藏门孜康制剂中心、神猴藏药厂等。阿如久巴丸即十味诃子丸,但为尊重传统用药习惯,在药品注册中由诃子、藏茜草、红花、刀豆、豆蔻、山矾叶、紫草茸、松蒂等十一味药味加工制成丸剂水丸,呈棕灰色至棕褐色,气芳香,味微酸、苦。具有清肾热、止痛、利尿等作用。临床上用于肾炎、泌尿生殖系统炎症及结石引起的尿频、尿急、尿痛、下腹部疼痛、腰膝酸痛、拖足跛行、下肢麻木等[1-2]。
本研究通过显微镜观察所收集到的制剂,能够准确描述制剂粉末的显微特征;采用薄层色谱法进行鉴别,对点样量、展开剂进行考察,确定最佳色谱条件;对检查项下的水分、灰分以及酸不溶性灰分等进行测定;并对浸出物含量进行了测定。本研究对具有阿如久巴丸生产批准文号的生产企业及制剂室进行样品和原料药材的收集,依次对收集的样品展开上述项目的质量标准研究,研究过程中参考了《中国药典》《中华人民共和国卫生部药品标准(藏药)》1995年版第一册以及十味诃子丸(阿如久巴丸)质量标准研究的文献报道[1,3,4]。拟定诃子、红花和藏茜草的显微鉴别;拟定诃子和红花药味的TLC鉴别。本研究对藏药阿如久巴丸质量控制提供了技术支持。
1 仪器与试药
1.1 仪器 正置光学显微镜(日本尼康,Nikon Eclipse E1000);电子天平(AL104型,瑞士METTLER-TOLEDO有限公司);紫外灯分析仪(ZF-I型,上海顾村电光仪器厂);薄层层析硅胶板:①硅胶G板(青岛海洋化工有限公司);②硅胶G板(烟台江友硅胶开发有限公司);③硅胶G板(青岛海浪硅胶干燥剂公司)。电热鼓风恒温干燥箱、马弗炉、可控调温电炉、电热恒温水浴锅等。
1.2 试药 本实验所使用的药材来自5个厂家共5个批次的样品,具体情况见表1。红花对照药材购自中国食品药品检定研究院(批号:110709-200803);诃子对照药材购自中国食品药品检定研究院(批号:121015-201605)。二甲苯、中性树胶、分析纯盐酸,超纯水、甲醇、无水乙醇、碘化铋钾、浓硫酸等所用试剂均是国产分析纯。

表1 阿如久巴丸企业信息及样品批号
2 方法与结果
2.1 显微鉴别 取5批阿如久巴丸样品粉末及按处方自配阿如久巴丸粉末2g,过4号筛。将过筛后的粉末用解剖针刮出均匀涂在载玻片上,滴加稀甘油封片,盖上盖玻片后分别在40×、100×、400×显微镜下观察。结果如图1所示。
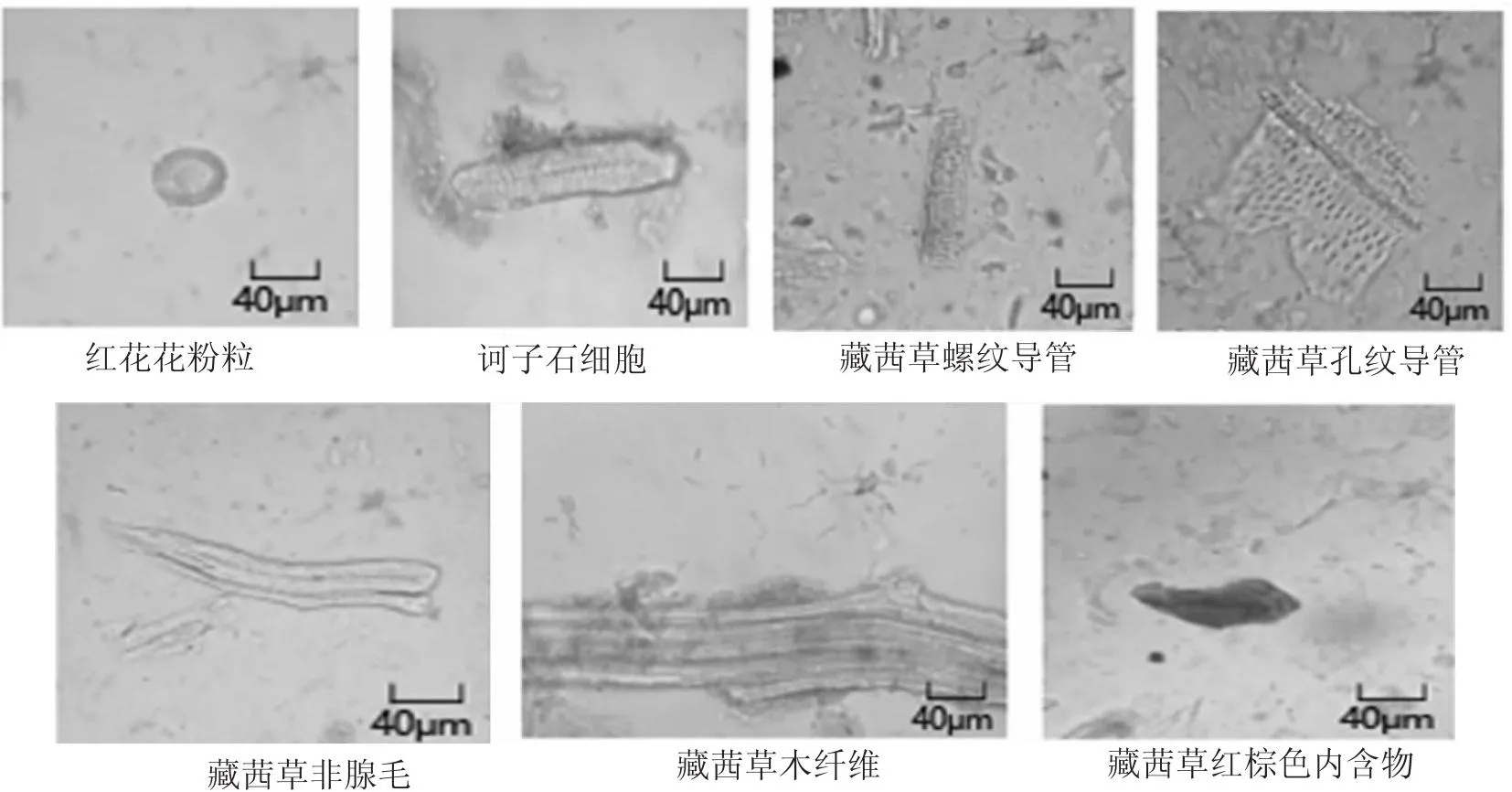
图1 阿如久巴丸粉末鉴定特征图
根据以上显微结果,标准叙述为:取本品,置显微镜下观察石细胞类方形、类多角形或呈纤维状,直径14~40 μm,长至130 μm,壁厚,孔沟细密(诃子)。有长管分泌细胞,含黄棕色至红棕色分泌物;花粉粒类圆形、椭圆形或橄榄形,直径约至60 μm,具3个萌发孔,外壁有齿状突起(红花)。非腺毛基部膨大,整体呈长圆锥状;木纤维孔纹明显;红棕色内含物多散在(藏茜草)。
2.2 薄层色谱鉴别
2.2.1 诃子薄层色谱鉴别[2-5]称取阿如久巴丸样品粉末2 g,置具塞锥形瓶中,加乙酸乙酯50 mL,超声处理20 min,滤过,滤液浓缩至1 mL,作为供试品溶液。按处方中各药味的比例,自配不含诃子的药材粉末作为阴性样品,按照供试品溶液的制备方法,制成阴性对照溶液。取诃子对照药材0.5 g,同供试品溶液制备方法制备对照药材溶液。将上述制备的供试品,阴性对照以及对照药材溶液用微量进样器点样于同一硅胶G薄层板,供试品溶液和阴性对照溶液各取10 μL,对照药材溶液3 μL,展开剂用三氯甲烷-丙酮-甲酸(7∶2∶1),待其预饱和20 min后,展开,取出,晾干,喷以2%三氯化铁乙醇溶液,105 ℃加热至斑点显色清晰。供试品色谱中,观察到各批次供试品溶液与对照药材溶液色谱相应的位置上显相同蓝色斑点,斑点清晰。3批供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。该鉴别方法对该药中诃子的鉴别具有很好的适应性,分离度好,Rf值适中,重现性较好。结果如图2所示。
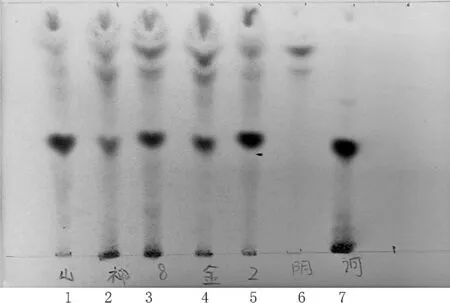
1~5.阿如久巴丸样品;6.诃子阴性对照;7.诃子对照药材图2 诃子薄层色谱鉴别图
本研究考察了3个展开系统,分别为三氯甲烷-乙醇-冰乙酸(6∶1∶0.1);三氯甲烷-乙酸乙酯-甲酸(5∶1∶0.2);三氯甲烷-丙酮-甲酸(7∶2∶1)。结果表明三氯甲烷-丙酮-甲酸(7∶2∶1)为展开系统时效果最好,斑点清晰,无拖尾,分离度良好,故以此为展开系统。同时本研究对点样量也进行了考察,对照药材溶液、供试品溶液点样量考察了3 μL、5 μL、10 μL,结果表明对照药材和供试品溶液分别为3 μL、10 μL时,斑点集中、清晰、不拖尾。故确定对照药材溶液、供试品溶液点样量分别为3 μL、10 μL。本研究对方法进行耐用性实验考察,采用不同厂家的薄层板考察:①硅胶G板(青岛海洋化工有限公司);②硅胶G板(烟台);③硅胶G板(青岛海浪硅胶干燥剂公司),展开剂为三氯甲烷-丙酮-甲酸(7∶2∶1),点样量为供试品溶液、对照品溶液各10 μL、3 μL,显色剂为2%三氯化铁乙醇溶液,于日光下检视。
结果表明,供试品在与对照药材色谱相应的位置上,显相同颜色的斑点。Rf值适中,分离度良好,斑点检视清晰,分别采用不同厂家生产的硅胶G板按照上述方法进行耐用性实验考察,结果在不同条件下该方法均能将样品良好的分离,斑点检视清晰,结果表明该方法耐用性良好。
2.2.2 红花薄层色谱鉴别[1-4]称取阿如久巴丸样品粉末3 g,置于具塞锥形瓶中,加入80%丙酮5 mL,密塞,振摇15 min,静置,取上清液作为供试品溶液。按处方中药材的比例,自配不含红花的药材粉末作为阴性样品,按照阿如久巴丸供试品溶液的制备方法,制成阴性对照溶液。取红花对照药材0.5 g,同供试品溶液制备方法制备对照药材溶液。将上述制备的供试品,阴性对照以及对照药材溶液用微量进样器点样于同一硅胶G薄层板,供试品溶液和阴性对照溶液,以及对照药材溶液各取5 μL,展开剂为乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),待预饱和20 min后,展开,取出,晾干。供试品色谱中,观察到各批次供试品溶液与对照药材溶液色谱相应的位置上显相同颜色的斑点,斑点清晰。3批供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。该鉴别方法对该药中红花的鉴别具有很好的适应性,分离度好,Rf值适中,重现性较好。结果如图3所示。
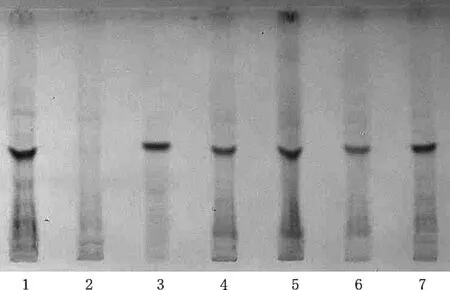
1.红花对照药材;2.红花阴性对照;3~7.阿如久巴丸样品图3 阿如久巴丸(红花)薄层鉴别图(18 ℃,45%RH)
本研究考察了3个展开系统,并对其比例进行调整,分别为氯仿-丙酮-冰乙酸(6∶1∶0.1)、氯仿-乙酸乙酯-甲酸(5∶2∶0.2)、乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),结果以乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4)为展开系统时效果最好,斑点清晰,无拖尾,分离度良好,故以此为展开条件。同时本研究对点样量也进行了考察,对照品溶液、供试品溶液点样量考察了3 μL、5 μL、10 μL,结果发现对照药材溶液和供试品溶液为5 μL时,斑点集中、清晰、不拖尾。故确定对照药材溶液、供试品溶液点样量分别为5 μL。本研究对方法进行耐用性实验考察,采用不同厂家的薄层板考察:①硅胶G板(青岛海洋化工有限公司);②硅胶G板(烟台);③硅胶G板(青岛海浪硅胶干燥剂公司),展开剂为乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),点样量为供试品溶液、对照品溶液各5μL,于日光下检视。
结果表明,供试品在与对照药材色谱相应的位置上,显相同的斑点。Rf值适中,分离度良好,斑点检视清晰,分别采用不同厂家生产的硅胶G板按照上述方法进行耐用性实验考察,结果在不同条件下该方法均能将样品良好的分离,斑点检视清晰,表明该方法耐用性良好。
2.3 阿如久巴丸水分含量、灰分及浸出物测定
2.3.1 水分测定 根据2020版《中国药典》四部通则0832水分测定项下[6],选取第二法烘干法对阿如久巴丸进行水分含量测定。
实验方法:差减法精密称取阿如久巴丸药材粉末3 g左右,平铺于干燥至恒重的已编号称量瓶中,厚度不超过5 mm,疏松阿如久巴丸药材粉末不超过10 mm,打开瓶盖在105 ℃下干燥5 h后将瓶盖盖好,移置干燥器中,冷却至室温,精密称定重量。再在上述温度干燥1 h,放冷,称重,至连续两次称重的差异不超过5 mg为止。累计烘干6 h。根据公式计算阿如久巴丸样品中含水量。
烘干法共测定5批阿如久巴丸,其中水分含量最高的批次为8.00%,最低为6.24%,平均值为6.99%。具体结果见表2。

表2 烘干法测定阿如久巴丸中水分含量结果表
实验结果表明,阿如久巴丸不同来源样品应用烘干法测定水分值相差最大在1.76%。按照2020版《中国药典》四部通则0832水分测定法测定,5批样品的水分均小于9.0%,符合规定。
2.3.2 灰分测定 根据《中国药典》2020版第四部通则2302灰分测定法[6],对阿如久巴丸样品进行总灰分及酸不溶性灰分的检测。
实验方法:差减法精密称取5 g阿如久巴丸样品于炽灼至恒重的已编号瓷坩埚中。在电炉上加热30 min使样品粉末碳化,待坩埚中无白烟冒出时表明样品粉末已碳化完全。将坩埚放入马弗炉中550 ℃下炽灼灰化2 h后取出,冷却至150 ℃左右放入干燥器中冷却至室温,称重,再放入马弗炉中灰化1 h,取出,称重。重复操作2次后达到恒重,累计炽灼4 h。根据公式计算阿如久巴丸中总灰分含量。
取上项所得的总灰分,在坩埚中加入10% 盐酸溶液10 mL,盖上盖子,置水浴上加热10 min,坩埚盖用5 mL热水冲洗,洗液并入坩埚中,用快速定量滤纸过滤,坩埚内的残渣用热水洗于滤纸上,并洗涤至洗液不显氯化物反应为止。滤渣连同滤纸移置同一坩埚中,同时在另一坩埚中放入一张快速定量滤纸作为空白实验。坩埚及样品105 ℃下干燥,电炉上加热至完全碳化后,移入马弗炉中550 ℃炽灼至恒重。根据公式计算阿如久巴丸样品中酸不溶性灰分的含量。
空白试验及5批样品结果见表3和表4,酸不溶性灰分空白试验结果如表3所示;阿如久巴丸的总灰分含量最高为23.10%,最低为19.87%,平均值为21.43%。试验结果见表4。

表3 酸不溶性灰分空白试验结果 (n=2)
实验结果表明阿如久巴丸的总灰分含量与酸不溶性灰分含量差异较大。推测阿如久巴丸中碱性无机成分含量较大的原因。各样品总灰分平均为21.43%,上浮20%为25.72%。酸不溶性灰分平均含量为0.296%,上浮20%为0.355%。对阿如久巴丸的总灰分及酸不溶性灰分的检测是保证阿如久巴丸用药安全的一项重要指标,建议总灰分不得过26.0%,酸不溶性灰分不得过0.36%,一批不合格。
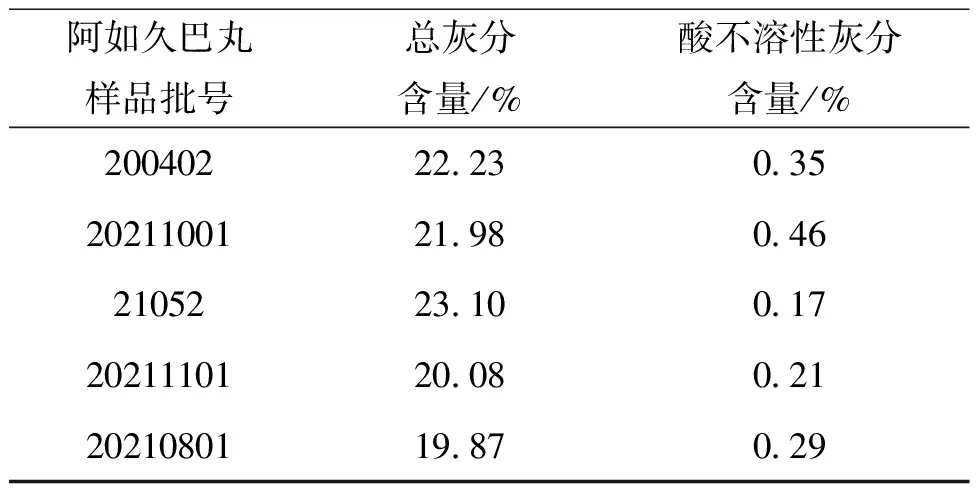
表4 阿如久巴丸的总灰分及酸不溶性灰分含量 (n=3)
2.2.3 浸出物测定 根据《中国药典》2020年版第四部通则2201浸出物测定法[6],测定阿如久巴丸浸出物含量。
实验方法:差减法精密称取4 g阿如久巴丸样品于250 mL锥形瓶中,精密加水100 mL,密塞,冷浸,前6 h内时时振摇,再静置18 h,用干燥滤器迅速滤过,精密量取续滤液20 mL,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105 ℃干燥3 h,置干燥器中冷却30 min,迅速精密称定重量。根据公式计算阿如久巴丸中浸出物的含量。
差减法精密称取5份3 g上述同一批次阿如久巴丸样品分别置于100 mL锥形瓶中,精密加水、30%乙醇、50%乙醇、70%乙醇及95%乙醇50 mL,密塞,称定重量,再静置1 h后,连接回流冷凝管,加热至沸腾,并保持微沸1 h。放冷后,取下锥形瓶,密塞,再称定重量,用所用溶剂补足减失的重量,摇匀,用干燥滤器滤过,精密量取续滤液25 mL,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105 ℃干燥3 h,置干燥器中冷却30 min,迅速精密称定重量。根据公式计算阿如久巴丸中浸出物的含量。
取同一批次的阿如久巴丸加同一溶剂95%乙醇,分别考察了冷浸法与热浸法,结果见表5。热浸法所得的浸出物含量高于冷浸法所得。故进而选择了热浸法对5种溶剂进行了考察,所得结果见表6。根据表6结果,最终选定95%乙醇为浸出物提取溶剂,对5个批次进行浸出物含量考察。结果见表7。

表5 阿如久巴丸浸出物的冷浸法与热浸法的考察 (n=3)

表6 阿如久巴丸浸出物提取溶剂的考察 (n=3)

表7 浸出物含量 (n=3)
实验结果表明来自西藏雄巴拉曲制药有限公司的阿如久巴丸的浸出物含量高,而来自西藏自治区藏医院门孜康制剂室阿如巴丸浸出物含量低,推测原因可能与原药材的产地、采收季节等因素有关,导致该批次的浸出物含量低。各批次浸出物含量平均值为33.35%。下浮20%为26.68%。对阿如久巴丸的浸出物含量检测是保证该药用药安全的一项重要指标,建议浸出物含量不得少于27.0%,有一批浸出物含量不合格。
3 结论
本研究基于《中华人民共和国卫生部药品标准(藏药)》1995年版第一册和中国药典基础上[3-4],通过显微鉴别实验,制定了阿如久巴丸中诃子、红花、藏茜草显微鉴别方法,通过薄层色谱实验,制定了阿如久巴丸中诃子与红花的薄层鉴别方法,完善了其质量标准,能有效帮助阿如久巴丸质量标准研究,为阿如久巴丸的质量标准建立奠定基础。
