Fe3+协同Ni基金属-有机框架提升电催化析氧反应活性的研究
2024-02-11陈星亮范文娟黄海平蒋志强
陈星亮,范文娟,常 会,黄海平,蒋志强
(1.江西理工大学材料冶金化学学部,江西赣州 341000;2.攀枝花学院钒钛资源综合利用四川省重点实验室,四川攀枝花 617000)
随着经济的快速发展,能源危机和环境污染已成为两大全球性问题[1]。开发可再生的清洁能源迫在眉睫[2-3],其中电解水被认为是最有前景的技术之一。电解水主要包括阴极析氢(HER)和阳极析氧(OER)两个半反应。阳极析氧反应涉及复杂的四电子转移过程,需要较高的过电位来克服动力学缓慢的问题,限制了电解水技术的实际应用[4]。因此,寻找一种高效的OER 催化剂,有效降低反应过电势和能耗成为了研究热点。贵金属Ru、Ir 基氧化物目前被认为是有效的OER 电催化剂,但由于其资源稀缺、价格昂贵,不适合实际应用。因此,开发廉价、高效且来源丰富的非贵金属OER 电催化剂具有十分重要的科学意义和应用价值。
金属有机框架(MOFs)是由中心金属离子与有机配体通过配位键形成的一类配合物,具有周期性多维网络结构[5-6]。该类材料不仅具有丰富美观的框架结构,而且由于其孔道尺寸、形状及主客体作用可调,在气体储存与分离、催化、能量转换和磁性等领域有着广泛的应用前景[6-10]。然而,MOFs材料固有的低稳定性极大地阻碍了其在电催化领域的进一步应用。尽管通过高温煅烧获得的MOFs衍生材料(如碳基材料、单原子掺杂材料等)被报道为高活性的电催化剂,但是MOFs 在高温下生成衍生材料是一个极其复杂的过程,结构难以实现精确控制,破坏了MOFs 材料本身独特的优势,且能耗较高[11-12]。因此,开发稳定高效的MOFs 基电催化剂具有十分重要的科学意义和应用价值。相关研究表明,在电解水过程中MOFs主要是利用框架内部分散良好的金属中心作为活性位点以提高催化活性[13-17]。其中,Ni-MOF由于含有较多的Ni活性中心,常表现出较好的电催化析氧性能,但是与商用RuO2、IrO2电催化剂相比,以Ni 为单一活性中心的Ni-MOF 的OER性能仍需进一步提高。由于Fe元素储量丰富,价格便宜,且相关文献研究表明Fe元素的掺杂可以显著提升电催化剂的催化活性,因此可通过Fe3+掺杂对Ni-MOF 进行改性,以此提升Ni-MOF 的电催化活性[18]。
本文首先采用溶剂热法制备出有机金属框架材料phen-Ni,然后在phen-Ni中引入异金属Fe3+,制备出Fe@phen-Ni催化剂。电催化析氧实验结果表明,Fe@phen-Ni 在0.1 mol/L KOH 电解液中表现出优异的催化活性和稳定性,并且在大电流密度下其OER性能显著优于商用电催化剂RuO2。
1 实验部分
1.1 药品及仪器
试剂:六水合硝酸镍[Ni(NO3)2·6H2O]、邻菲啰啉(phen)、4,4'-二羧基二苯醚(H2oba)、三乙胺、硝酸铁、无水乙醇、硫酸、氢氧化钾、RuO2,均为分析纯;Nafion(全氟化树脂,质量分数为5%)溶液、T 型泡沫镍,均购自于上海河森电气有限公司。
仪器:FA220413 型电子分析天平、DHG-9036A型电热恒温鼓风干燥箱、15D044型超声波清洗机。
1.2 电催化剂的制备
金属有机框架材料phen-Ni的具体制备方法参考文献[19]。将Ni(NO3)2·6H2O (0.145 2 g,0.5 mmol)、邻菲啰啉(0.091 8 g,0.5 mmol)、4,4'-二羧基二苯醚(0.129 7 g,0.5 mmol)、三乙胺(0.150 2 g,1 mmol)和去离子水(10 mL)依次加入到23 mL 聚四氟乙烯高压反应釜中,超声30 min 后放入160 ℃烘箱,反应72 h后取出,自然冷却至室温,抽滤并使用去离子水和乙醇洗涤3次,经真空干燥后得到绿色块状晶体,标记为phen-Ni。称取100 mg phen-Ni加入到20 mL 0.1 g/mL硝酸铁的乙醇溶液中,室温下浸泡24 h,过滤并经真空干燥后得到Fe@phen-Ni。
1.3 工作电极的制备
将电催化剂phen-Ni、Fe@phen-Ni、RuO2分别置于玛瑙研钵中,研磨1 h。称取10 mg研磨后的电催化剂于1.5 mL 离心管中,依次加入50 μL Nafion 溶液、100 μL无水乙醇和350 μL超纯水,超声30 min,制得分散均匀的悬浮液。移取40 μL 悬浮液均匀涂覆于T型泡沫镍(NF)正反两面(0.5 cm×1 cm),自然风干后得到工作电极,分别记为phen-Ni/NF、Fe@phen-Ni/NF、RuO2/NF,其中催化剂负载量为1.6 mg/cm2。
1.4 催化剂表征方法
通过D8 Advance 型X 射线单晶衍射仪(PXRD)分析晶体结构;使用DX-2007 型X 射线粉末衍射仪(XRD)分析材料物相;使用Inspect F50 型扫描电子显微镜(SEM)观察电催化剂的微观形貌;采用Escalab250 Xi 型X 射线光电子能谱仪(XPS)测试电催化剂的元素组成和价态;利用JEOL JEM LaB6 2100型透射电子显微镜(TEM)、高分辨透射电子显微镜(HRTEM)及能谱分析仪(EDS)表征电催化剂的微观结构;借助VERTEX 70V 型傅里叶变换红外光谱仪获得电催化剂中化学键或官能团信息。
1.5 电催化活性测试
采用三电极体系利用上海辰华电化学工作站(CHI 760E)进行电催化活性评价。实验在N2饱和的0.1 mol/L KOH溶液中进行,phen-Ni/NF、Fe@phen-Ni/NF、RuO2/NF 作为工作电极,铂片作为对电极,Ag/AgCl 作为参比电极,盐桥为3 mol/L KCl。首先,通过循环伏安法(CV,0~0.8 V,50 mV/s)对催化剂进行活化,直到CV曲线完全重合,以去除电催化剂表面污染物。然后,在扫描速率为5 mV/s 条件下使用线性扫描伏安法(LSV)评估OER 催化活性,在0.6 V(vs.Ag/AgCl)、频率范围为0.02~1×106Hz 条件下测试电化学阻抗(EIS)。电化学活性面积(ECSA)与催化剂的双电层电容值(Cdl)呈正比,选取电位区间0.830 3~0.930 3 V[vs.可逆氢电极(RHE)],以不同扫描速率(20~60 mV/s)获得CV曲线,并以此计算出Cdl值。采用计时电流法进行电化学稳定性测试,在0.66 V(vs.Ag/AgCl)下循环测试8 h 后研究电流衰减情况。实验中所有的电流(A)归一化为电流密度(mA/cm2),并根据方程ERHE=EAg/AgCl+0.059 1 pH+0.197,将测得的电位转换为可逆氢电极电势。
2 实验结果与讨论
2.1 晶体结构
室温下通过PXRD 以2θ扫描方式收集衍射数据,并采用SAINT 程序还原。所有结构通过SHELXL和Olex2程序并采用直接法进行解析,利用全矩阵最小二乘法对结构进行精修。表1 为phen-Ni的主要晶体学参数。

表1 phen-Ni的主要晶体学参数Table 1 The main crystallographic parameters of phen-Ni
由表1 可知,phen-Ni 属于单斜晶系,具有P21/n空间群。图1 为phen-Ni 中Ni2+的配位环境。由图1a 可知,phen-Ni 最小不对称单元包含1 个Ni(Ⅱ)、1个去质子化的oba2-配体、1个phen配体和1个游离的H2O。其中,六配位的金属Ni 中心与来自2 个不同oba2-配体的4个氧原子(O1a、O2a、O4、O5)及1个phen 配体的2 个氮原子(N1 和N2)配位。Ni—O和Ni—N 键长分别为[2.064(3)~2.216(4)]Å 和[2.089(4)~2.096(4)] Å。两个金属Ni 中心通过去质子化的oba2-配体连接形成类似“Z形”的链式结构(见图1b),由此形成的链式结构采用π-π堆积的方式组成三维网络结构(见图1c)。

图1 phen-Ni的Ni2+配位环境Fig.1 Coordination environment of Ni2+ in phen-Ni
2.2 表征结果
在水溶液体系中进行电催化反应时,要求催化剂必须具有良好的水稳定性,尤其应具有优异的耐强酸、强碱能力。图2 为样品的PXRD 谱图。由图2a 可知,制备的phen-Ni 的PXRD 谱图与Mercury 软件导出的模拟谱图吻合,说明获得了单一物相组成的phen-Ni 材料。将phen-Ni 置于不同pH 的溶液(H2SO4溶液、KOH 溶液)中浸泡24 h,研究其酸碱稳定性,结果如图2b 所示。由图2b 可知,phen-Ni 在不同pH 溶液中浸泡后的PXRD 谱图与浸泡前的谱图基本一致,表明phen-Ni能在pH为3~13的酸碱环境中保持稳定。根据上述酸碱稳定性测试结果,选取pH=13 的KOH 溶液作为电解质,评价其电催化活性。

图2 样品的PXRD谱图Fig.2 PXRD patterns of samples
对掺杂前后的材料进行红外光谱表征,结果如图3 所示。由图3 可知,在2 691~3 709 cm-1处的宽峰为phen-Ni 骨架中结合水的O—H 伸缩振动峰;在1 117、1 242、1 643 cm-1处的吸收峰分别对应于4-4'-二羧基二苯醚结构中的C—O—C、C—O、C=O伸缩振动峰;在1 405、1 608 cm-1处的吸收峰分别对应于邻菲啰啉结构中的C—N和C=N伸缩振动峰;在474 cm-1处的吸收峰对应于Ni—O 伸缩振动峰,613 cm-1处的吸收峰对应于Ni—N 伸缩振动峰,776 cm-1处的吸收峰对应于苯环上C—H 弯曲振动峰。以上结果说明Ni与邻菲啰啉和4,4'-二羧基二苯醚成功配位,该结果与PXRD 测试结果一致。经对比发现,phen-Ni 与Fe@phen-Ni 的特征吸收峰一致,没有多余的吸收峰产生,表明Fe元素在phen-Ni中不是以键合的方式存在,而是通过离子交换封存在phen-Ni的骨架中。
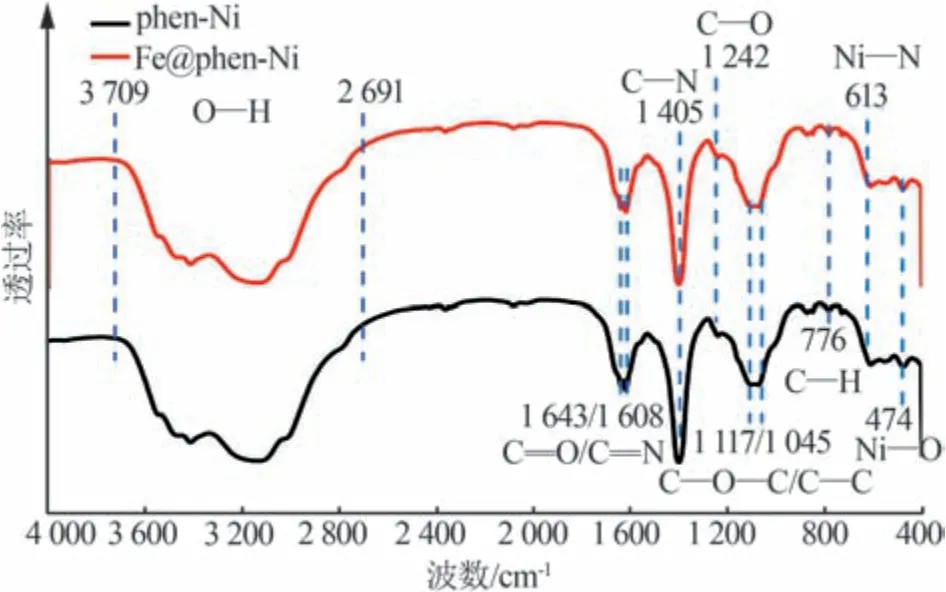
图3 phen-Ni和Fe@phen-Ni的FT-IR谱图Fig.3 FT-IR spectra of phen-Ni and Fe@phen-Ni
图4为Fe@phen-Ni的SEM图。从图4可以看出,Fe@phen-Ni呈棒状结构,长约500 nm,宽约200 nm。利用透射电子显微镜和高分辨透射电子显微镜进一步对Fe@phen-Ni 的微观结构进行表征,结果如图5所示。从图5 可以看出,Fe@phen-Ni 呈棒状结构,与SEM 结果一致。Fe@phen-Ni 的EDS 测试结果显示Ni、Fe、C、N均匀分布在材料中,表明Fe元素均匀掺杂到phen-Ni中(图5d1~d4)。

图4 Fe@phen-Ni的SEM图Fig.4 SEM images of Fe@phen-Ni

图5 Fe@phen-Ni的TEM图(a、b)、HRTEM图(c)及元素分布图(d1~d4)Fig.5 TEM images(a,b),HRTEM image(c),and element distribution images(d1~d4) of Fe@phen-Ni
利用XPS 表征了Fe@phen-Ni 的元素组成和价态,结果如图6所示。由图6a可知,在Ni 2p谱图中,结合能为873.6 eV 和855.9 eV 处的峰分别归属于Ni 2p1/2和Ni 2p3/2,邻近的两个峰为Ni 2p1/2和Ni 2p3/2的卫星峰,表明材料中的Ni 是以Ni2+的形式存在。在Fe 2p谱图(图6b)中,结合能为724.8、711.7 eV处的峰分别归属于Fe 2p1/2和Fe 2p3/2,表明材料中存在Fe3+。由图6c可知,在C 1s谱图中,结合能为284.7、285.1、286.1、291.8 eV处的峰分别归属于C—C=C、C—N、C—O—C、O=C—O。在N 1s 谱图(图6d)中,位于399.9 eV 处的峰归属于吡咯氮。XPS 结果进一步证实,Fe3+成功掺入到phen-Ni 材料中,这与TEM测试结果一致。Fe3+提供了丰富的电催化活性位点,有利于电子的转移,可显著提升Fe@phen-Ni的OER性能。

图6 Fe@phen-Ni中Ni 2p(a)、Fe 2p(b)、C 1s(c)和N 1s(d)高分辨XPS谱图Fig.6 XPS high-resolution spectra of Ni 2p (a),Fe 2p (b),C 1s (c) and N 1s (d) of Fe@phen-Ni
2.3 催化剂的OER电催化活性
图7 a为催化剂的极化曲线。基于1.5节转换方程,结合图7a 得出,当电流密度为10 mA/cm2时,Fe@phen-Ni/NF、RuO2/NF、phen-Ni/NF 和NF的过电位依次为333、343、418、425 mV,Fe@phen-Ni/NF 的过电位略低于RuO2/NF;当电流密度达到50 mA/cm2时,Fe@phen-Ni/NF 的过电位(410 mV)明显低于RuO2/NF(441 mV),这得益于Fe3+与phen-Ni 中Ni2+的协同作用,提升了phen-Ni 的OER 电催化活性。NF和phen-Ni表现出较差的OER电催化能力,表明Fe@phen-Ni/NF 较高的OER 催化活性归因于Fe@phen-Ni,Fe3+的掺杂不仅增加了催化活性位点,而且异金属位点的存在使反应环境更加丰富,改变了金属位点的电子结构,从而提升电催化剂的OER活性。此外,Fe@phen-Ni/NF 的过电位低于许多已报道的MOF 基电催化剂,如CTGU-14(376 mV)[20]、NNU-23(365 mV)[21]、NCNTFs(370 mV)[22]、CoOx-ZIF (400 mV)[23]。

图7 催化剂的OER催化性能Fig.7 OER electrocatalytic performance of catalysts
为了进一步得到电催化材料的电催化动力学参数和速率控速步骤,可将LSV 曲线通过Tafel 关系式[η=blgj+a(η为过电位,b为Tafel斜率,j为电流密度,a为依赖于电极材料的常数)]换算获得Tafel斜率,结果如图7b 所示。由图7b 可知,Fe@phen-Ni/NF 的Tafel 斜率为105.5 mV/dec,低于NF(177.9 mV/dec)、phen-Ni/NF(169.7 mV/dec)和RuO2/NF(119.2 mV/dec),表明OER 过程中Fe@phen-Ni/NF表现出更优异的动力学行为。通过CV 曲线测定双层电容值(Cdl),得到催化剂催化过程中实际的电化学活性面积(ESCA),Cdl与电化学活性面积成正比。图7c 为电流密度与扫描速率的线性关系。由图7c可知,Fe@phen-Ni/NF 的Cdl值(2.4 mF/cm2)大于NF的Cdl值(0.92 mF/cm2),表明Fe3+的引入提供了更多的活性位点,利于水分子的吸附及与电解液的接触等,从而提高了电化学活性面积,提升了电催化活性。较低的Cdl值对应于较低的过电位和Tafel斜率,揭示Fe@phen-Ni/NF优异的电子传输能力和更快的催化动力学与极化曲线和Tafel斜率结果一致。
NF、RuO2/NF、phen-Ni/NF 和Fe@phen-Ni/NF 在频率为0.02~1×106Hz、电压为0.6 V(vs.Ag/AgCl)条件下的电化学阻抗谱如图8a 所示。由图8a 可知,Fe@phen-Ni/NF 的电荷转移电阻Rct最小,表明Fe@phen-Ni/NF在电解质界面上的电子转移速度最快。为了进一步评价Fe@phen-Ni的电催化稳定性,在0.66 V(vs.Ag/AgCl)电压下通过恒压计时电流法(i-t)对Fe@phen-Ni/NF 进行了8 h 的稳定性测试,结果如图8b 所示。由图8b 可知,i-t曲线虽然略有下降,但整体平稳,这可能是因为长时间的析氧反应测试产生的O2附着在工作电极表面,阻碍了催化剂与电解液的充分接触。图8c 为Fe@phen-Ni/NF 在稳定性测试前后的极化曲线。由图8c 可知,Fe@phen-Ni/NF在稳定性测试前后的极化曲线几乎完全重合,进一步证实i-t曲线的轻微下降并非是由活性下降引起的,因此Fe@phen-Ni/NF在KOH(pH=13)溶液中具有优异的电催化OER稳定性。
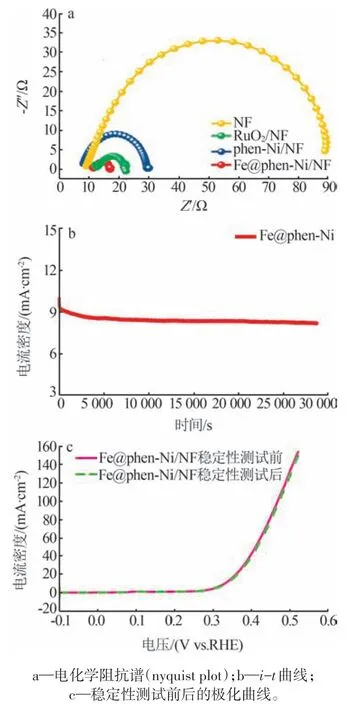
图8 催化剂的OER阻抗及稳定性测试Fig.8 OER impedance and stability test of catalysts
3 结论
1)本文通过溶剂热法合成了phen-Ni,然后通过Fe3+掺杂,制备得到Fe@phen-Ni 电催化剂。2)在0.1 mol/L KOH 电解液中,Fe@phen-Ni 相较于phen-Ni 和RuO2显示出更为优异的OER 催化活性和稳定性。3)Fe@phen-Ni 优异的电催化活性主要归因于Fe3+和Ni2+的协同作用,异金属位点的存在使局部反应环境更加丰富,改变了金属位点的电子结构,提高了本征催化剂的活性,为MOFs 基电解水OER 催化剂的制备提供了一种新的策略。
