病毒感染与宿主抗感染免疫之间“博弈”
——凋亡、坏死和焦亡分子机制
2024-02-01陈松彪刘飞飞余祖华张春杰程相朝
陈松彪,刘飞飞,尚 珂,余祖华,何 雷,魏 颖,陈 建, 张春杰,程相朝*,丁 轲*
(1.洛阳市活载体生物材料与动物疫病防控重点实验室,洛阳 471003;2.河南科技大学功能微生物与畜禽健康 实验室,洛阳 471003;3.动物病原与生物安全教育部重点实验室,郑州 450000)
病毒感染首先与宿主细胞表面受体相结合进入细胞,病毒感染细胞后迅速“占领”宿主领地并将其改装成为病毒加工制造“工厂”,其目的就是在此处大量合成产生新的感染性病毒粒子[1]。宿主控制和消除病毒复制传播的有效方法之一就是通过程序性细胞死亡消除感染细胞,尽管有报道称病毒在复制过程中诱导应激导致细胞发生死亡,有助于病毒在感染后期释放子代病毒粒子。但细胞早期死亡也诱导宿主形成了强大抗病毒防御机制:(1)细胞死亡破坏了病毒自身加工制造“工厂”;(2)细胞死亡大量释放胞内危险相关分子模式(damage associated molecular patterns,DAMPs)来促进宿主固有免疫反应;(3)细胞死亡促进了树突状细胞对病毒抗原的摄取和呈递,促进宿主适应性免疫进程[2-3]。宿主抗病毒感染免疫三种重要的细胞死亡途径分别是凋亡(apoptosis)、坏死(necroptosis)和焦亡(pyroptosis),不同死亡模式由不同的特异信号级联分子驱动[4-5],这些细胞死亡途径启动不同细胞形态学的解体过程。
许多病毒感染会导致宿主细胞的细胞膜失去完整性,从而有利于合成子代病毒释放,病毒持续性感染对正常细胞功能的损害最终会导致细胞死亡。比如腺病毒E1A蛋白,它通过诱导细胞周期DNA复制期和有丝分裂期调节剂关键酶pRb(视网膜母细胞瘤抑制蛋白)失活,迫使宿主细胞复制周期进入DNA合成的S期(有丝分裂间期),以大量合成脱氧核苷酸来促进新病毒基因组DNA生成,在此过程中也诱导p53激活来促使内源性死亡受体通路的细胞凋亡[6];与此相反,在此过程中病毒所编码的E1B-55K蛋白还能够通过与p53结合,来阻断其促凋亡活性[7]。越来越多的证据显示,病毒诱导宿主细胞死亡蛋白和拮抗宿主细胞死亡蛋白活性存在时间/空间上存活-死亡之间相对平衡[8-9]。近年来研究也发现病毒和宿主之间通过不同“博弈”来进行感染和免疫,其中细胞凋亡、坏死和焦亡在此过程中发挥了重要作用。因此,本论文主要综述了细胞凋亡、细胞坏死和细胞焦亡在病毒感染与宿主抗感染免疫过程中的作用及调控机制,以期能为深入研究病毒致病机制以及开发新的抗病毒药物提供参考。
1 病毒感染与宿主抗感染免疫“博弈”——细胞凋亡
1.1 细胞凋亡
细胞凋亡又称“细胞程序性免疫沉默”[10],是由半胱氨酸天冬氨酸蛋白酶(cysteinyl aspartate specific proteinase,Caspase)家族成员介导去除有害或者异常细胞等而产生细胞自我调节过程,即主动程序性细胞死亡方式[11]。当病毒进入细胞后,宿主模式识别受体(pattern recognition receptors, PRRs)对其基因组感知引发细胞凋亡是对宿主发挥先天免疫反应抗病毒感染重要组成部分,病毒基因组可以以多种形式被感知,包括病毒DNA(vDNA)、病毒RNA合成的cDNA(v-cDNA)和病毒单链或双链RNA(vRNA)。细胞凋亡受级联反应信号控制,并通过两个重要信号通路发生:外源性的死亡受体通路(extrinsic death receptor pathway)和内源性的线粒体通路(intrinsic mitochondrial pathway)[12-14]。
1.1.1 外源性死亡受体通路 外源性途径是由细胞外的信号通过死亡配体如肿瘤坏死因子(tumor necrosis factor,TNF)、Fas配体(fas ligand,FasL)和肿瘤坏死因子相关的凋亡诱导配体(TNF-related apoptosis-inducing ligand,TRAIL)与各自的受体结合而启动的[15]。死亡配体和相应受体结合后募集Fas相关死亡结构域(fas associated death domain,FADD)、Caspase-8前体(Pro-Caspase8)形成死亡诱导信号复合体(death inducing signaling complex,DISC),通过同型相互作用导致Caspase-8激活,激活的Caspase-8可直接水解激活效应蛋白“凋亡执行者”Caspase-7和Caspase-3诱导细胞凋亡[16](图1)。
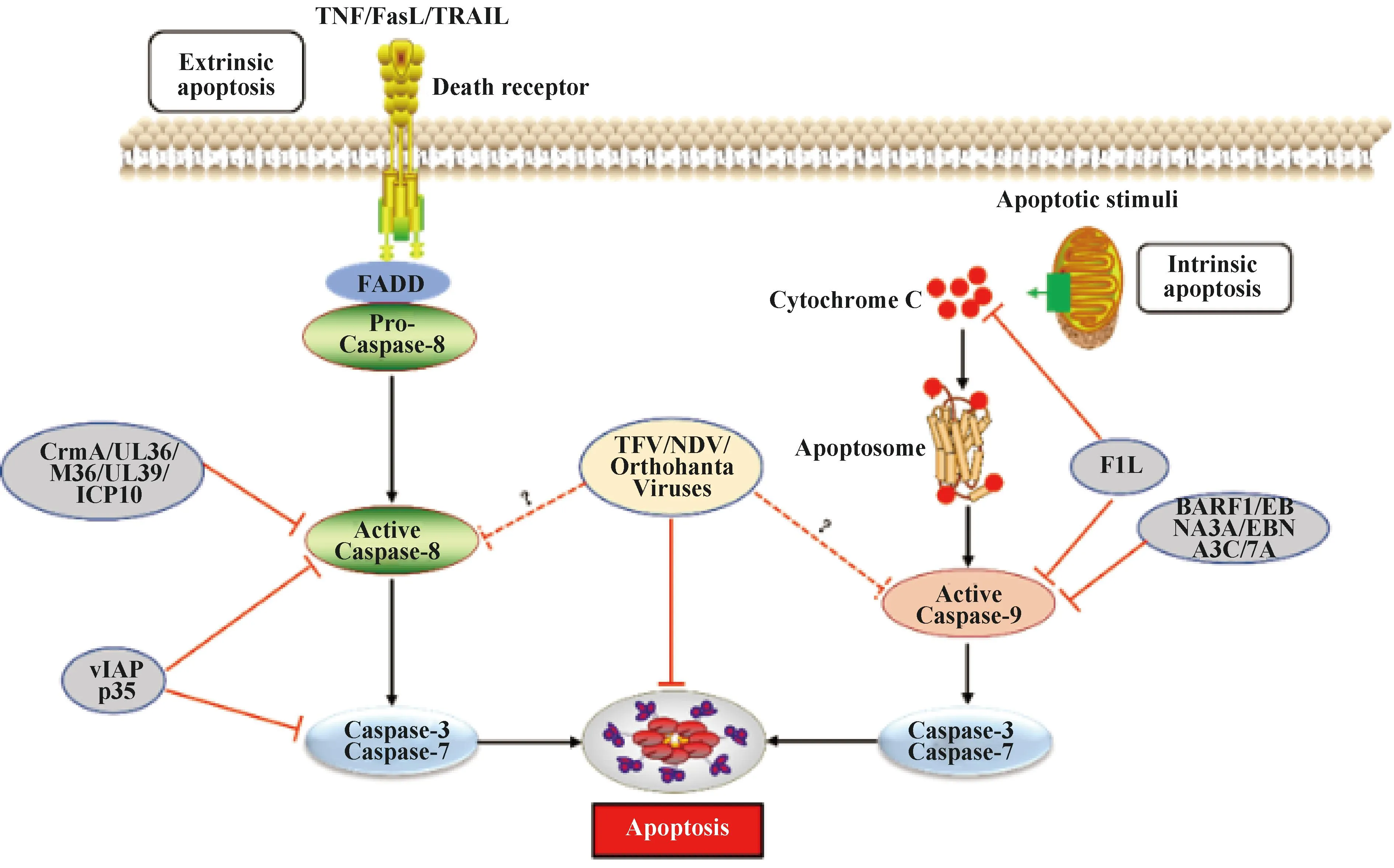
图1 病毒感染与宿主抗感染免疫“博弈”——细胞凋亡Fig.1 Virus infection and host antiviral immunity struggle-cell apoptosis
1.1.2 内源性线粒体通路 与外源性细胞凋亡不同,内源性途径是被DNA损伤、存活因子缺失、生长因子阻断或其它细胞内应激等激活,引起Bcl-2家族促凋亡蛋白(Bax、Bak、Bad、Bid、Puma、Bim和Noxa等)和抗凋亡蛋白(Bcl-2、Bcl-xl、Bcl-w和Mcl-1等)表达发生变化,然后促凋亡蛋白诱导线粒体外膜通透性(mitochondrial outer membrane permeabilization,MOMP)增加,从而导致细胞色素C(cytochrome C)从细胞器的膜间空间释放,游离细胞色素C与Apaf-1结合形成凋亡小体复合物(apoptosome),该复合物进一步激活Caspase-9来剪切下游的“凋亡执行者”Caspases-3和Caspases-7诱导凋亡[17](图1)。病毒感染可通过上调Bcl-2家族中促凋亡蛋白诱导细胞凋亡。人类免疫缺陷病毒Ⅰ型(HIV-1)感染能够上调Puma和Bax水平[18-19];发热伴血小板减少综合征病毒[20]、哺乳动物呼肠孤病毒[21]、塔卡里伯病毒[22]以及丙型肝炎病毒[23]等感染上调Bak、Noxa或者Bik等促凋亡因子水平。同时,病毒感染也能够下调Mcl-1或(和)Bcl-2等抗凋亡蛋白来诱导凋亡[24-25]。病毒感染诱导细胞凋亡表现为细胞有序解体和发生特定形态学变化[26],包括细胞皱缩、质膜完整、细胞质致密、细胞器密集、核裂解、细胞质多发性芽突,并迅速脱落形成凋亡小体,这些凋亡小体被吞噬细胞吞噬后清除。
1.2 病毒感染抑制宿主细胞凋亡的分子机制
1.2.1 病毒感染抑制外源性死亡受体介导的细胞凋亡途径 细胞凋亡是宿主抗病毒感染免疫的一种重要手段,病毒为了自身有效复制进化出各种不同复杂机制来抵制宿主细胞凋亡。第一个由病毒自身编码的凋亡抑制分子是牛痘病毒(Vaccinia virus,VACV)细胞因子修饰剂A(CrmA),CrmA具有广泛的丝氨酸和半胱氨酸蛋白酶抑制活性,能够有效抑制Caspase-1、Caspase-8和Caspase-9的活性[27],后续在其它痘病毒中也相继发现CrmA的同源蛋白[28](图1)。病毒凋亡抑制蛋白(vIAPs)是另一类广谱Caspase抑制剂,vIAPs同源物在杆状病毒感染的蝴蝶、飞蛾和苍蝇中被发现,大多数病毒和细胞IAPs包含具有泛素连接酶活性的杆状病毒IAP重复序列和C末端的环指结构域(really interesting new gene,RING),迄今为止发现超过200种vIAPs都是由感染昆虫的大型DNA病毒所携带。多角体病毒vIAP的p35可作为人类Caspase-1、Caspase-3、Caspase-6、Caspase-7、Caspase-8和Caspase-10活化的抑制剂[29],进一步结构信息学分析发现p35可以和底物竞争性结合Caspase-8催化位点来抑制其活性,但是其如何抑制Caspase-1、Caspase-3、Caspase-6、Caspase-7和Caspase-10活性尚不清楚[30](图1)。人巨细胞病毒(HCMV)、小鼠巨细胞病毒(MCMV)、单纯疱疹病毒1型(HSV-1)以及HSV-2基因组编码UL36[31]、M36[32]、UL39[33]以及ICP10[34]蛋白,能够与Caspase-8相互作用,抑制其蛋白水解酶活性(图1)。此外,γ-疱疹病毒和软疣病毒属成员表达c-FLIP(vFLP)同源物,一种类似于缺少Caspase-8催化活性蛋白酶结构域的短c-FLIP亚型,c-FLIP与Caspase-8形成异质二聚体,导致Caspase-8失活来阻断其介导的细胞凋亡作用,从而阻断凋亡信号通路[35-36]。
1.2.2 病毒感染抑制内源性线粒体通路介导的细胞凋亡途径 病毒感染后宿主可通过上调Bcl-2家族中促凋亡蛋白或下调抗凋亡蛋白诱导细胞凋亡,从而阻止病毒的复制[37]。但是,病毒在长期进化过程中利用表达自身病毒蛋白也可以通过相应策略来抑制凋亡[38]。EB病毒(Epstein-Barr virus,EBV)编码的BARF1蛋白通过上调病毒感染细胞中抗凋亡蛋白Bcl-2和Bcl-xl的表达水平来抑制细胞凋亡[39],同时其编码的EBNA3A和EBNA3C蛋白可下调促凋亡蛋白Bim的表达水平,抑制细胞凋亡[40](图1)。乙型肝炎病毒(HBV)上调非编码RNA lnc-HUR1来抑制p53转录活性,进而上调凋亡抑制因子Bcl-2、抑制凋亡促进因子Bax转录来抑制细胞凋亡[41]。此外,VACV、严重急性呼吸综合征冠状病毒(SARS-CoV)以及风疹病毒(RV)等病毒可编码蛋白通过与Bcl-2家族蛋白相互作用影响其正常功能抑制细胞凋亡[42-44]。VACV编码F1L蛋白能够结合Bcl-2家族蛋白,抑制细胞色素C释放,从而抑制Caspase-9的激活。进一步研究显示F1L也可直接利用其N端结构域与Caspase-9互作来抑制其活性[43],从而抑制由过表达Caspase-9所诱导凋亡[44](图1)。RV衣壳蛋白通过直接与Bax结合抑制细胞凋亡,防止Bax激活诱导细胞凋亡[45]。对轮状病毒属[46]、副黏病毒属[47]和布尼病毒属[48]等成员研究也发现病毒感染过程中采取不同策略抑制细胞凋亡从而促进自身复制,但具体作用机制尚不清楚。
2 病毒感染和宿主抗感染免疫“博弈”——细胞坏死
2.1 细胞坏死
由于外界不利因素引起细胞正常代谢活动受损或者发生中断时,在受体相互作用蛋白激酶1(receptor-interacting protein kinase 1,RIPK1)及其下游的RIPK3和混合谱系激酶域样蛋白(mixed lineage kinase domain-like,MLKL)将会激活细胞程序性坏死通路。细胞程序性坏死是一种新型的细胞炎性死亡方式,以胞质内蛋白形成坏死小体,细胞膜打孔破裂释放内容物诱导炎症为主要特征。不同于细胞凋亡,坏死和焦亡都不参与维持细胞正常生理机能,而是以溶解和炎症反应为特征细胞死亡方式,具有重要抗病毒功能[49]。
细胞表面肿瘤坏死因子(tumor necrosis factor α,TNFα)受体1((tumor necrosis factor receptor 1,TNFR1)是死亡受体超家族的一员,其刺激可激活程序性坏死通路[49],TNFα与TNFR1结合形成由TNFR1相关的死亡结构域蛋白 (TNFR1-associated death domain protein,TRADD)、TNF受体2(TNF receptor associated factor 2,TRAF2)、细胞内凋亡蛋白质抑制蛋白1/2(cIAP1/2)和RIPK1组成的膜信号复合物I(Complex I),随后激活NF-κB通路从而促进细胞存活和炎症形成(图2)。当RIPK1泛素化被抑制时Complex I将被释放并募集FADD,形成TRADD-FADD-Caspase-8组成复合物II(Complex II α),进而激活凋亡信号通路。在病毒感染期间或对Caspase-8活性进行药物抑制,RIPK1和RIPK3借助它们的RIP同源基序互作结构域(RIP homotypic interaction motif,RHIM)发生结合形成坏死小体,坏死小体启动RIPK3介导的MLKL磷酸化,磷酸化的MLKL破坏质膜并导致细胞坏死[50](图2)。值得注意的是,在大多数生理条件下,RIPK1-RIPK3的细胞坏死复合体形成会受到cFLIP/Caspase-8异质二聚体复合物抑制,该复合物通过接头蛋白FADD被募集到RIPK1-RIPK3复合物中,并能同时降解RIPK1和RIPK3以防止坏死和促进凋亡[51]。因此,Caspase-8活性的抑制(类似于MCMV表达vICA蛋白功能)是诱导细胞坏死的关键步骤。
近年来研究发现,细胞坏死也可以被胞内感受器Z-DNA结合蛋白1(ZBP1)激活,当病毒释放了核酸进入细胞内,ZBP1能够识别并结合细胞质中的双链DNA分子,同时激活RIPK3,使其磷酸化,获得激酶活性,激活的RIPK3磷酸化MLKL,诱导细胞坏死(图2)。在流感病毒和传染性法氏囊病毒复制过程中产生Z-RNA(一种新的PAMP分子),这些Z-RNA激活感染核中的ZBP1,ZBP1被激活后刺激RIPK3,RIPK3发生磷酸化并激活细胞核中的MLKL,MLKL引发核膜破坏,促进细胞DNA释放至细胞质中,从而诱导细胞坏死(图2)[52-53]。
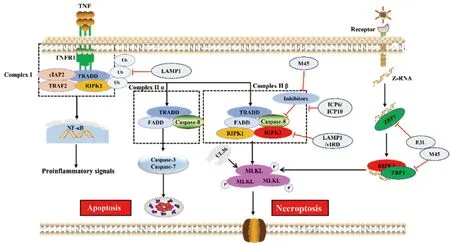
图2 病毒感染与宿主抗感染免疫“博弈”——细胞坏死(参照文献[53]并进行修改)Fig.2 Virus infection and host antiviral immunity struggle-cell necroptosis (Modified according to reference[53])
2.2 病毒感染抑制宿主细胞坏死的分子机制
病毒针对细胞这一特性,进化出各种复杂“武器”阻止细胞坏死这一自杀性行为,从而实现复制和生存。主要靶向于细胞坏死通路中的关键分子MLKL、RIPK1/RIPK3和ZBP1。
2.2.1 靶向于MLKL抑制细胞坏死 疱疹病毒家族多个成员编码含有RHIM结构域蛋白,干扰宿主细胞RIPK3组装以及伴随的下游MLKL激活。MCMV、HSV-1、HSV-2基因组编码M45[54]、ICP6[55]、ICP10[34]蛋白通过依赖性N末端RHIM结构域与RIPK1、TRIF以及ZBP1互作,竞争性抑制Complex II β复合体形成,减弱MLKL磷酸化从而抑制细胞坏死(图2)。MCMV编码具有酶活性UL36蛋白,与MLKL互作通过蛋白酶体途径促进MLKL降解,从而抑制坏死[31]。痘苗病毒(vaccinia virus,VV)编码vIRD蛋白能够促进RIPK3泛素化和蛋白酶体降解途径来抑制下游MLKL磷酸化来抑制细胞坏死[8]。
2.2.2 靶向于RIPK1和RIPK3抑制细胞坏死 牛痘病毒、鼠痘病毒和天花病毒均能表达另一种具有降解RIPK3功能的病毒蛋白vIRD,在病毒感染过程中vIRD通过其N端重复序列与RIPK3结合,通过其C端F-box与细胞SKP1-CUL1复合体结合促进RIPK3的K48型链泛素化和蛋白酶体介导的降解,抑制感染细胞发生坏死[56]。EBV表达LMP1通过两个不同途径抑制坏死:干扰RIPK1和RIPK3泛素化以及降低RIPK3表达[57]。在痘病毒中还发现病毒编码的蛋白COTV157和BAV Rmi具有和MLKL同源的假激酶结构域(缺乏活性激酶的大多数或所有催化基序),通过它们和RIPK3互作竞争性阻止MLKL结合和磷酸化,从而抑制坏死[58]。
2.2.3 靶向于ZBP1抑制细胞坏死 VACV所编码病毒蛋白E3L,该蛋白包含两个不同的双链RNA(dsRNA)结合结构域,这两个结构域都对毒性至关重要:E3L蛋白N-末端含有Z-核酸结合域(Zα),它专门与Z-RNA结合,C-末端包含一个典型的dsRNA结合结构域,它与典型的α构象中的dsRNA相互作用。Zα结构域与ZBP1的功能结构域相似,能够隔离Z-RNA的配体,阻止ZBP1的活化来抑制细胞坏死从而逃避宿主天然免疫反应[59]。此外,MCMV病毒蛋白M45含有RHIM结构域,可以干扰ZBP1和RIPK3之间的相互作用抑制细胞坏死(图2)[60]。以上报道表明坏死不仅发生在Caspase-8活性被阻断的情况下,而且可能作为一种单独的机制来抑制病毒感染,但是凋亡-坏死之间切换信号及相应调控机制仍不清楚。
3 病毒感染和宿主抗感染免疫“博弈”——细胞焦亡
3.1 细胞焦亡
细胞焦亡是细胞感染时由炎症小体介导,以裂解细胞为特点的程序性死亡形式,是机体重要的天然免疫反应,在拮抗和清除病原菌感染中发挥关键作用[61-62]。细胞焦亡激活信号途径分为依赖Caspase-1的经典途径和依赖Caspase-4、Caspase-5或Caspase-11的非经典途径,这两种途径均通过切割炎性半胱氨酸蛋白酶D(gasdermin D,GSDMD),释放出N端游离的肽段,该肽段会诱使细胞形成孔道导致细胞破裂发生焦亡。
3.1.1 依赖Caspase-1的信号通路 依赖Caspase-1的细胞焦亡通路又称为经典炎症小体信号通路。炎症小体是天然免疫系统的重要组成部分,能够识别并激活病原菌/微生物相关分子模式(pathogen-associated molecular patterns,PAMPs/DAMPs),招募和激活促炎症蛋白酶Caspase-1,其中最常见的炎症小体是NOD样受体蛋白(NLR family pyrin domain containing 1,NLRP1、NLRP3和NLRC4)和黑色素瘤缺乏因子2(absent in melanoma 2,AIM2)样受体[4]。炎症小体是一个多蛋白复合物,其激活通常是由PRRs与含有半胱天冬酶募集结构域(caspase activation and recruitment domain,CARD)的凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing a caspase-recruitment domain,ASC)之间的同质相互作用诱导引发,ASC通过CARD-CARD相互作用募集并激活Caspase-1,Caspase-1活化后切割IL-1β和IL-18前体,形成成熟促炎细胞因子IL-1β和IL-18[63]。活化的Caspase-1也可以切割GSDMD的N端和C端结构域的连接区,释放具有结合膜磷脂上膜打孔活性的N端结构域,导致细胞质膜上形成孔,随后细胞膜破裂,释放大量炎症细胞因子和DAMPs,引发细胞焦亡[9](图3)。
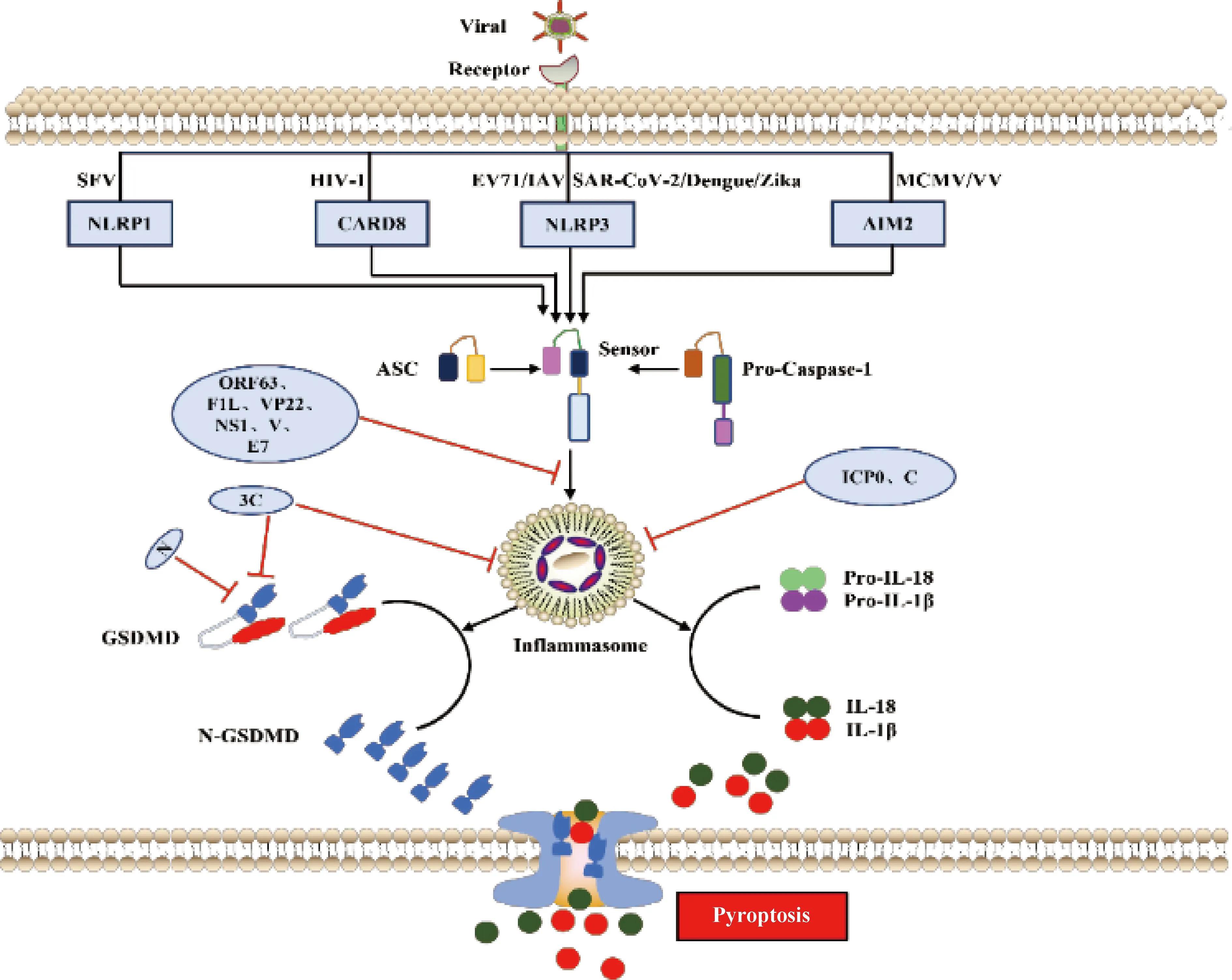
图3 病毒感染与宿主抗感染免疫“博弈”——细胞焦亡(参照文献[10]并加以修改)Fig.3 Virus infection and host antiviral immunity struggle-cell pyroptosis (Modified according to reference[10])
NLRP1在塞姆利基森林病毒(SFV)感染或合成的dsRNA模拟物poly(I:C)转染过程中充当dsRNA传感器,NLRP1通过其富含亮氨酸的重复结构域与dsRNA相互作用,诱导NLRP1寡聚以及激活Caspase-1形成炎症小体,最终促使焦亡发生[64];登革病毒(Dengue virus)[65]、寨卡病毒(Zika virus)[66]、肠病毒(EV)[67]以及新型冠状病毒(SARS-CoV-2)[68]感染后利用不同的手段促进NLRP3表达发生上调,促进Capase-1的活化和炎症小体形成来诱导焦亡;HIV-1感染利用CARD8主要作为HIV-1蛋白酶感受器,CARD8在其感染后诱导细胞焦亡过程中发挥重要作用[69];同时发现MCMV[70]和VV[71]感染后,通过AIM2识别并激活下游Caspase-1来诱导细胞焦亡。
3.1.2 依赖Caspase-4、Caspase-5或Caspase-11的信号通路 依赖Caspase-4、Caspase-5或Caspase-11信号通路又称Caspase-1非依赖细胞通路或非经典炎症小体通路,该信号通路是由人源Caspase-4和Caspase-5或鼠源Caspase-11触发,这些Caspases活化后切割GSDMD(和Caspase-1相同切割位点)并启动细胞焦亡[72-73](图3未显示)。近些年对非经典通路炎症小体通路研究较少,下文主要阐述病毒抑制经典通路之细胞焦亡。
3.2 病毒感染抑制宿主细胞焦亡的分子机制
虽然病毒感染过程中有许多炎症小体和焦亡激活的例子,但病毒也可以通过调节炎症小体激活或靶向焦亡“执行者”GSDMD直接抑制细胞焦亡来促进自身复制。
3.2.1 DNA病毒抑制宿主细胞焦亡 卡波齐肉瘤疱疹病毒编码NLRP1同源物蛋白ORF63, ORF63可竞争性抑制NLRP1炎症小体活化以及下游的Caspase-1介导的细胞焦亡[74]。VV基因组编码vBcl2蛋白同源物F1L具有双重功能,F1L体外结合并抑制NLRP1,缺乏F1L病毒感染巨噬细胞后导致Caspase-1激活和IL-1β分泌增加。缺失F1L造成病毒毒力在体内减弱,引起发热反应改变和Caspase-1水解增加,同时伴随感染小鼠肺部炎症加速但不影响细胞死亡或病毒复制[75]。HSV-1可拮抗AIM2和IFI16介导的炎症小体激活,HSV-1蛋白VP22与AIM2炎症小体相互作用,阻止其寡聚化,从而抑制AIM2活化,而ICP0泛素连接酶活性则靶向IFI16,使其通过蛋白酶体途径发生降解[76-77]。人乳头瘤病毒E7蛋白通过促进TRIM21介导的IFI16炎症小体泛素化降解抑制细胞焦亡[78]。兔黏液瘤病毒和兔纤维瘤病毒表达病毒pyrin样蛋白,这些蛋白与接头蛋白ASC直接相互作用以抑制NLRP3炎症小体组装[79-80](图3)。
3.2.2 RNA病毒抑制宿主细胞焦亡 除了DNA病毒抑制宿主细胞焦亡促进自身复制外,一些RNA病毒也通过不同策略来抑制细胞焦亡。柯萨奇病毒和脑心肌炎病毒编码3C蛋白酶可切割NLRP3和GSDMD使其失活,但NLRP1被相同的病毒蛋白酶裂解导致N-甘氨酸特异性降解,导致NLRP1活化[62,81]。人NLRP1和小鼠同源蛋白NLRP1B的病毒蛋白酶切割位点进化成病毒多聚蛋白的切割位点,这表明NLRP1蛋白与肠道病毒蛋白酶共同进化来发挥宿主抗病毒感染[80]。甲型流感病毒的NS1以及副黏病毒家族成员麻疹病毒、仙台病毒和尼帕病毒的V蛋白可阻断NLRP3炎症小体的激活[82-83](图3)。人副流感病毒3型利用C蛋白通过蛋白酶体途径降解NLRP3来阻止炎症小体激活[84](图3)。此外,SARS-CoV-2编码N核衣壳蛋白通过阻止Caspase-1介导的切割和GSDMD的激活来阻断人单核细胞焦亡和IL-1β分泌[85-86](图3)。综上所述,病毒感染宿主细胞后引发炎症小体组装激活是一个相当复杂的生物学过程,宿主细胞通过细胞质内的感受器第一时间识别病毒的不同组分,如结构蛋白和病毒核酸等,并产生相应的炎症小体进而激活后续炎症和免疫相关反应导致细胞焦亡,从而抑制病毒感染和复制。同时,病毒可以利用合成自身编码蛋白来抑制炎症小体的组装和激活,以逃避宿主细胞抗感染免疫。
4 小结与展望
病毒进化出不同策略来拮抗感染过程中细胞凋亡、坏死和焦亡,从而促进了自身复制。病毒成功逃避了宿主重要的抗感染免疫途径——细胞死亡,可能会诱导出宿主细胞新的替代抗感染免疫途径出现。同时当细胞受到病原微生物感染刺激时,细胞是否由于处于不同生理状态而导致一些细胞对病原微生物刺激“抵抗”从而发生凋亡、坏死或焦亡?这些细胞选择性抗感染免疫信号和调控机制又是什么?
目前研究发现病毒感染和宿主抗感染免疫之间的博弈类型也存在着时间和空间上关联。例如,猪细小病毒感染过程中能够发生凋亡、自噬性死亡(完全自噬和不完全自噬的转化)[87-90],在RNA病毒小反刍兽疫病毒感染过程中也发现存在第一波自噬流和第二波自噬流[91],然而这些不同通路之间互相切换的信号及其确切机制仍不清楚。另外,还有研究发现颗粒酶A和颗粒酶B能够激活GSDMB和GSDME,从而引起肿瘤细胞死亡[92-93],那么以颗粒酶A/GSDMB和颗粒酶B/GSDME细胞死亡轴为基础开发出小分子模拟物是否能够临床应用清除肿瘤感染细胞?随着标签蛋白和同位素示踪在病毒学研究中广泛应用,使得人们能够更清楚、更全面认识在病毒感染不同时期细胞微环境所发生的不同类型变化。未来的研究可通过活细胞实时成像、病毒示踪以及病毒反向遗传操作技术综合分析,来阐明病毒操纵宿主细胞死亡以及宿主抗感染免疫的分子机制,将为进一步揭示病毒的致病机制和合理设计精准靶点来选择性干扰宿主细胞死亡的病毒拮抗剂提供新的思路。
