关于实测分解电压低于可逆分解电压的讨论*
2024-01-25何荣桓南松波
何荣桓,陈 帅,南松波
(东北大学理学院化学系,辽宁 沈阳 110819)
依据热力学原理,化学反应分为可自发进行和不可自发进行两种。对于不能自发进行的化学反应,通过外加能量对体系做功有可能促使反应的发生,例如,设计电池外加一个直流电源,当外加电源电压增加到一定数值时就会在电极上发生化学反应,此过程称为电解。发生电极反应所需的电压即为分解电压(E分解),由理论分解电压和过电位组成。
由于过电位的存在,实际分解电压通常高于理论分解电压,而实际分解电压往往由实验测得,如图1所示。
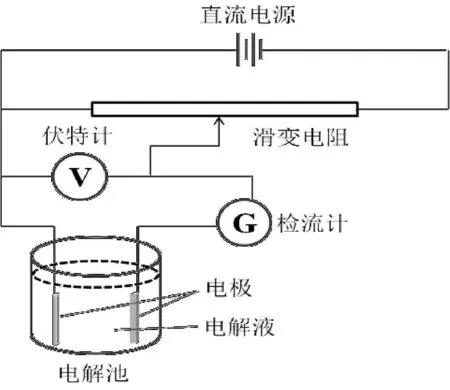
图1 分解电压的测定示意图
电解伊始,外加电压较小几乎没有电流通过。当外加电压逐渐增大时,电极表面微量产物与电极构成原电池,产生与分解电压相反的反电动势Eb(back electromotive force);随着电压逐渐增加并克服反电动势,电解产物逐渐增多并开始向溶液扩散,Eb不再增加而电流呈缓慢增加(图2,1→2段);此时继续增大外加电压,离子定向移动产生溶液中的电位降 (IR=E外-Eb)。当外加电压增加到某一数值以后,曲线的斜率明显增大,电流就会随电压增大而呈直线上升趋势(图2,2→3段),记录电流随电压的变化曲线,便可得到如图1所示的曲线。将图中直线部分3外延到电流强度为零时横坐标所对应的电压就是分解电压E分解。理论上讲,E分解对应原电池的可逆电动势E可逆,然而测量中实际发生的不可逆电解过程致使E分解>E可逆。
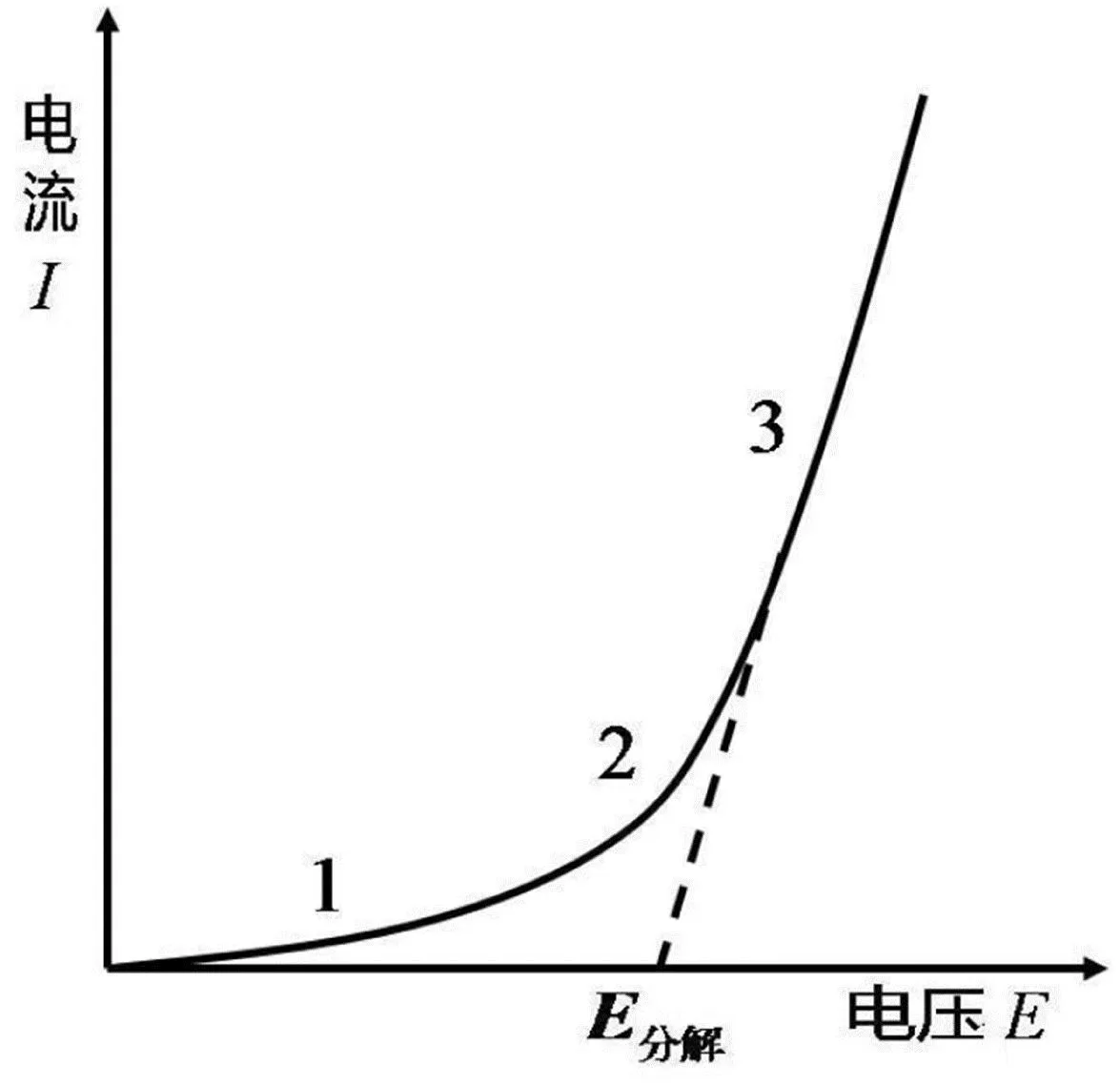
图2 电解过程中的电流-电压曲线
1 问题的提出
表1给出了几种电解质水溶液的分解电压和与可逆电动势的比较结果。然而在对表1的数据进行观察分析后我们发现了如下问题:

表1 几种电解质水溶液的分解电压(298 K,100 kPa,以一价离子计,浓度为1 mol·dm-3)[1]
(1)未指明金属的具体存在状态,例如金属单质是否以汞齐形式存在,其对应的标准电极电位会有所不同,但表1数据未标记金属的存在状态,因此难以严格对应所列出的可逆分解电压E可逆;
(2)对于以盐为电解质的体系,当体系的酸碱度不确定时,即使电解生成物相同,也会导致可逆分解电压数值的不确定性,难以进行E分解与E可逆的比较;
(3)表1虽然列出了E分解-E可逆的差值,但对于相关结果缺乏相应的分析讨论,尤其是实测分解电压低于可逆电动势的这一反常情况,给读者带来了困扰;
(4)由于上述(1)和(2)问题的存在,也使得问题(3)的解决缺乏精确的定量依据。
2 结果与讨论
电解过程中影响实际(或实测)分解电压的因素很多,考虑表1已有的实验条件和分析讨论的可行性基础,我们对相关影响因素的讨论作以下假定和说明:
(1)表1数据界定了温度、压力、电解质组成及浓度、以及具体电解产物等条件,以下讨论以表中结果科学可靠为基础展开,并不再赘述这些因素的变化产生的影响;
(2)由于电解池构成、电极种类、电极表面状态、以及实验测量误差的不可知性,假定由这些因素导致的不同数据组之间可能存在的合理误差可以忽略不计;
(3)本文的分析讨论聚焦于实测分解电压低于理论分解电压这一“反常”现象,因此着重分析电化学过程中不可抗拒或难以避免的客观存在和现象等因素对结果的影响,例如,过电位,欠电位沉积及溶解吸附等。
2.1 电极反应及物态
由于表1援引教材中没有标注数据出处,所以无法搜寻确定具体实验条件。针对上述情况,我们利用标准电极电位(298.15 K,101.325 kPa),本着与表1所提供数据最接近的情况,对有关物态和介质酸碱性进行了匹配;匹配数据附在原数据后面的括号“()”中以示区别(结果见表2);在此基础上,以原表提供的实测分解电压与可逆电动势进行比较,对比较结果进行了进一步的分析讨论。

表2 几种电解质水溶液的分解电压及电极反应 (以一价离子计,浓度为1 mol·dm-3)[1-3]
2.2 电解过程及影响因素分析
2.2.1 过电位
标准电极电位通常是在理想可逆状态下的电位,而实际进行的过程均为不可逆过程,通常都偏离可逆状态,因此存在偏离平衡可逆状态的过电位在所难免,只是偏离程度视具体反应和实验条件不同而不同。一般而言,有气体析出的电极反应通常都具有较大的过电位,具体机理比较复杂,因此至今尚不是很清楚。首先表1和表2中,除了以ZnBr2为电解质的体系外,其余所有水电解体系均有至少一种气体(H2,或O2,或Cl2)产生。尽管电极过程和相关机理迄今依然有待研究,但气体析出通常需要经历多个步骤已经被人们普遍接受,包括:相关物种从溶液本体扩散至电极表面→在电极表面吸附发生电荷转移→原子化合生成气体→生产的气体从电极表面脱附等。其中多个步骤中哪个是决速步骤需根据具体情况进行深入研究分析。由于存在迟缓放电和多步复合过程,因此涉及气体生成的反应除了电极活化极化(actvition overpotential)之外,还会有浓差极化(concentration overpotential),具体到电池系统,引起过电位的还可能会有电解质离子传质阻力,阴极和阳极物质相互渗透导致混合电位,多孔电极中的传质损失,以及内部少许电子流等导致的偏离等。因此同样电流密度下,有气体生成的电化学反应通常都有较高的过电位,所以表中大多数实测E分解高于E可逆。也就是说,因过电位导致的实测分解电压只会高于理论分解电压而不是出现相反结果,故利用过电位解释不了表1中的反常现象。
2.2.2 电解质中离子的相互作用(活度因子)
从表1数据可以看出,对于有卤素单质生成的体系,E分解与E可逆的差值普遍较低,在0.21~-0.14 V范围,且明显低于没有卤素产物的体系(最低为0.44 V);当电解质为HCl,HBr,HI,和ZrBr2时,出现了E分解低于E可逆的“异常”情况;此外,同样有Cl2生成的CoCl2与NiCl2为电解质的电解体系,E分解与E可逆之差分别为0.09 V和0.21 V,虽然差值明显低于其他没有卤素单质生成的体系,但未出现负值。因此,我们将分析讨论集中在有卤素(X)单质X2生成的HX为电解质的体系。
电极反应:
2X--2e=X2(正极);2H++2e=H2(负极)
根据能斯特方程有:
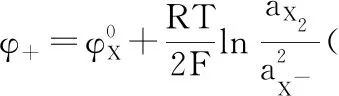
体系电压应为:
从上式可以看出,参与反应的各物质的活度可能影响E分解与E可逆的差值。对于单质产物来说,其初始活度远小于可逆状态单质的活度,由此可能导致欠电位沉积发生,此问题放在下一节讨论。对于HX电解质,其浓度为定值,因离子之间相互作用导致电解质活度下降,由此会引起实测分解电压升高,所以电解质活度因子不是导致体系E分解低于E可逆“异常”的原因。
2.2.3 欠电位沉积
欠电位沉积(underpotential deposition,UPD)是指一种物质(例如金属)发生单分子层电沉积时的电位明显偏离热力学可逆电位的现象,对于金属来说,其发生欠沉积沉积的电位通常比可逆电位更正。一般认为这是由于沉积原子之间的相互作用弱于基底与沉积原子之间的相互作用而造成的。对于有卤素离子X-存在的电池体系,一方面,其很容易在某些金属表面发展欠电位吸附而被氧化,导致在偏离热力学可逆电位更负下析出单质X2;另一方面,卤素阴离子X-的存在影响金属的欠电位沉积[4]。例如,卤素阴离子X-(F-除外)存在时会在金电极表面形成Cu-X吸附层,有利于金属的欠电位沉积[5]。有研究指出,卤素阴离子X-的存在对金属在金(111)电极表面形成吸附层M-X的影响顺序为Cl-< Br-< I-;然而对于金属Zn在金(111)电极上的沉积,上述影响顺序则不存在,I-的存在甚至抑制金属 Zn 的欠电位沉积[6]。综上分析可以看出,卤素阴离子X-的存在对欠电位沉积的确存在较大影响,然而其影响的电位的变化方向存在一定的不确定性。
2.2.4 电解产物的溶解度与反电动势
鉴于具体电解过程机理的复杂性难以在此企及,我们将本着把问题简单化对上述结果进行讨论说明。

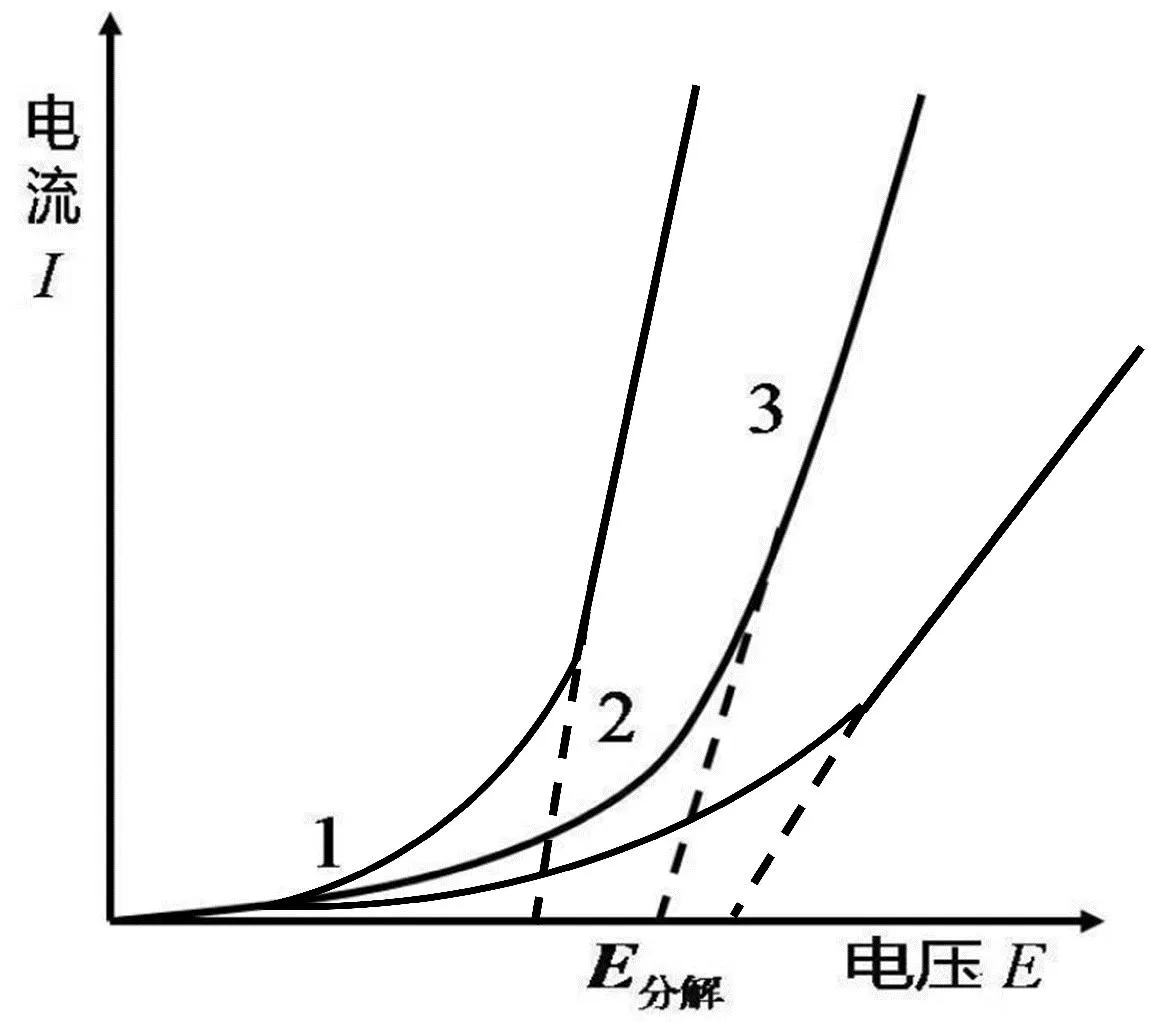
图3 不同电解质电解过程中电流-电压示意图

表3 几种电解产物常压下在水中的溶解度*(数字右上角为温度/℃)[3]
进一步地,电解反应的发生与电极与电解质之间的界面性质密切相关,比如,很多金属的还原析出电位高于本体析出电位(对应活度为1时的可逆电位)。对于有卤素氯、溴和碘生成的氧化过程:
2HX=H2+X2
卤素单质的平衡析出电位应为:
电解伊始,aX2< 1,且卤素单质的溶解度越大,其aX2越小,导致实际发生电解的电位φ’小于对应本体溶液的φe,即实际发生电解的电位低于平衡电位,使得E分解变小,出现了E分解与E可逆的差值为负值的情况。
根据上述分析,我们将各种可能的影响因素及该影响导致的电位变化情况汇总于表4中。可见,导致实测分解电压出现反常现象的原因主要是由欠电位沉积和卤素单质在水中溶解度较大、而反电动势较小造成的,而后者有着较大的影响。

表4 影响分解电压测定的因素汇总使((E分解-E可逆)结果增大记为“+”,反之为“-”,不确定记为“±”)[1]
需要指出的是,此处所讨论的数据来自教材,由于其所用电极以及电解具体条件无从得知,无法进行进一步的深入分析,因此所有分析讨论局限于表中有限的数据结果,不涉及电极种类、电极状态、以及电解池的组装等具体条件。另一方面,具有线性关系的3的斜率为I/V,与电解液的电导密切相关,其外延截距决定了E分解的大小,因此说,电解液导电能力从某种程度也影响截距的数值,鉴于对应电解质的实际电导率无从得知,此处不作讨论。
3 结 语
一般来说,电解池的分解电压大于可逆电动势。当电解产物中有卤素单质生成,且析出的金属过电位很小可忽略不计时,出现了实测分解电压低于可逆电动势的情况,通过对影响因素的分析讨论,指出出现实测分解电压反常的情况除了欠电位沉积因素外,主要是由于生成的卤素单质在水中溶解度较大,需对抗的反电动势小,电解过程较易发生而导致的。由于影响电解过程的因素除了电极和电解质种类、电极状态、生成产物等因素外,系统的组装等也会有较大的影响,由于无法获知更多实验条件,此处的讨论无法涵盖这些因素的影响。
