木质素还原制备金纳米颗粒及其催化性能
2024-01-25李茉琰龙杏张清桐梁展明闵斗勇
李茉琰,龙杏,张清桐,梁展明,闵斗勇
(广西大学轻工与食品工程学院, 广西清洁化制浆造纸与污染控制重点实验室, 南宁 530004)
木质素是芳香族化合物中少有的天然可再生资源之一,其含量仅次于纤维素,广泛存在于植物细胞中。在制浆企业生产过程中,作为工业废弃物的木质素大部分在碱回收工段进行燃料处理获得蒸汽或热能[1],仅有 5%的工业木质素用于制造分散剂、添加剂、表面活性剂和胶黏剂等高价值商业产品,大部分都没有得到合理的利用[2-3]。木质素分子结构中还原性的酚羟基可作为还原剂,将金属离子还原成金属单质并生长成纳米颗粒。柠檬酸钠是最早被用于合成金属纳米粒子的还原剂,由于柠檬酸钠还原性弱,用这种方法制备出的纳米粒子单分散性较差且纳米粒子尺寸难以调控[4]。相较于水合肼(H6N2O)、硼氢化钠(NaBH4)等有毒还原剂,木质素可以在不添加其他外源还原剂的条件下将Pt2+、Ag+、Au3+等金属离子还原并生成金属纳米粒子[5]。这种利用木质素的还原性制备贵金属纳米粒子的方法,为开发一种新型木质纤维素高值化利用手段提供了新的切入点。
金(Au)是化学性质最稳定的元素之一,而纳米级别的金具有独特的光电、物化性质及良好的生物相容性[6];因此,在众多的纳米材料研究中,金纳米颗粒(Au NPs)是被研究最多、最广泛以及最深入的纳米材料之一。Au NPs在构建生物传感器、研究电化学催化、光电、物化性能等方面都有很广阔的应用前景。Au NPs具有高比表面积,活泼的表面键态、电子态,表面电子配位不全等特点,赋予了其独特的催化活性,是催化还原染料废水中亚甲基蓝(MB)或难生化降解废水中对硝基苯酚(4-NP)的优良催化剂。作为典型的有机污染物,MB与4-NP广泛存在于纺织、造纸、印刷等工业废水中,因其具有较强的光稳定性和抗氧化性,很难被生物降解,对人体健康和自然环境构成严重威胁[5,7-8]。Wu等[9]对Au NPs催化NaBH4还原4-NP的性能展开了研究,结果表明Au NPs具有优异的催化活性,但其高比面积和高表面能导致其稳定性差,在水溶液中极易发生聚沉,导致催化活性显著降低。目前已报道了多种在载体上沉积Au NPs的方法,如光沉积、化学沉积,浸渍和化学还原等,但这些技术中的大多数在控制成本和简化实验步骤等方面受到限制。此外,采用传统工艺得到的Au NPs通常具有多分散性,这是由于金属纳米颗粒之间的重力沉降和捕获引起的前驱体溶液分散不均匀导致的。而木质素的三维网状结构可以有效阻止Au NPs在水溶液中发生聚沉[10-11]。
笔者以木质素为还原剂,在太阳光驱动下将Au3+还原成Au (0)并进一步生长成Au NPs,替代传统制备方法中所用的水合肼、NaBH4等有毒试剂[12],深入探究了Au3+物质的量浓度、木质素质量分数、光照时间等不同变量对Au NPs尺寸和形貌的影响;再以MB和4-NP为模拟污染物,探究Au NPs/木质素的催化性能,揭示其光催化机理,为木质素高值化利用提供一定的理论依据。
1 材料与方法
1.1 试验材料
酶解木质素由山东龙力科技股份有限公司提供;4-NP、MB、NaBH4和HAuCl4购自上海阿拉丁生化科技股份有限公司,均为分析纯;其他试剂均为国产分析纯。
1.2 酶解木质素纯化
酶解木质素纯化步骤如下:1)称取30 g酶解木质素,室温条件下,溶解在100 mL 1 mol/L NaOH中,过滤除去不溶杂质; 2)在滤液中逐滴加入10%(质量分数) HCl,直至溶液pH=2;3)在4 000 r/min 下离心10 min,收集沉淀物并用去离子水重复多次洗涤,冷冻干燥获得粗木质素;4)称取10 g粗木质素溶于100 mL丙酮,连续搅拌6 h,使木质素均匀溶解在丙酮中。然后在4 000 r/min的转速下离心10 min,收集上层清液,35 ℃下旋蒸,冷冻干燥获得纯化木质素。
1.3 木质素结构表征
采用AGILENT 1260型凝胶渗透色谱(GPC,美国安捷伦)分析纯化木质素样品的分子量。取50 mg木质素与1 mL吡啶充分混合后再加入1 mL乙酸酐混合均匀,室温下避光反应48 h,经乙醇和去离子水洗涤并旋转蒸发3次后,得到乙酰化木质素。使用不同相对分子质量的聚苯乙烯建立标准曲线,测试乙酰化木质素的数均分子量(Mn)、重均分子量(Mw)和多分散性(PD)。
采用AVANCE Ⅲ HD600型31P核磁共振(31P-NMR,德国Bruker)测定纯化木质素样品的羟基官能团含量。将0.4 mmol乙酰丙酮铬加入10 mL氘代吡啶中,再加入5 mmol N-羟基琥珀酰亚胺溶液作为内标,充分溶解后即得到含有弛豫剂的内标溶液;取15 mg木质素样品置于5 mL棕色样品瓶中,再依次加入400 μL 二甲基亚砜(DMSO)、200 μL 内标溶液和80 mL 2-氯-4,4,5,5-四甲基-1,3,2-二氧磷杂环戊烷(TMDP),震荡摇匀至样品完全溶解后立即转移到核磁管中进行测试(测试参数:脉冲角90°,温度298 K,扫描次数32次,谱图宽度100×10-6,等待时间6 s,中心频率145),各级分官能团含量计算公式如下[13]:
酚羟基的含量=A/A′×0.01/w
(1)
式中:A为样品中酚羟基的积分面积;A′为内标量N-羟基琥珀酰亚胺的积分面积;0.01为样品溶液中31P NMR的物质的量,mmol;w为样品质量,g。
1.4 金纳米颗粒/木质素分散体系制备
称取10 mg纯化木质素溶于100 mL丙酮,搅拌30 min得到0.1 g/L木质素溶液。按一定体积比将木质素溶液与HAuCl4溶液均匀混合,在模拟太阳光(500 W氙灯)下反应1 h,得到Au NPs/木质素悬浮液。HAuCl4浓度分别为0.05,0.10,0.50,1.00,1.50,2.00,2.50和3.00 mmol/L,HAuCl4溶液与木质素溶液(0.1 g/L)的体积比分别为1∶10,1∶2,1∶1,2∶1,3∶1,4∶1和5∶1,光照时间分别为3,5,10,20,30,60,90,120,150,180和210 min。
1.5 Au NPs/木质素性质表征
采用Ruli HT7700型透射电子显微镜(TEM,日本日立)观察Au NPs的形貌;采用ZS90X型纳米粒度仪(美国Malvern Panalytial)测定Au NPs的粒径与分布;采用SPECOND PLUS 50型紫外-可见光谱仪(UV-Vis,德国Analytik Jena)测定Au NPs悬浮液的紫外-可见光谱;采用THERMO ESCALAB 250Ⅺ 型X射线光电子能谱仪(XPS,美国Thermo Fisher)分析样品组分与化合态(单色Al Ka X射线(1 486.68 eV),扫描范围为0.0~1 350.0 eV,通过能为30 eV,扫描数为3次)。
1.6 催化性能测定
利用去离子水分别配制100 mL 0.1 mmol/L MB和0.1 mmol/L 4-NP。为防止NaBH4分解,在冰水浴中配制100 mL 40 mmol/L NaBH4。分别将1.5 mL NaBH4溶液与1.5 mL MB、4-NP在标准比色皿(光程长度为1 cm)中混合,加入100 μL Au NPs/木质素溶液,利用UV-Vis (测量模式为光谱扫描,扫描范围为200~800 nm,Delta为2 nm,速度为200 nm/s)分别在664和400 nm处监测MB和4-NP特征峰的变化。加入不同浓度的Au NPs/木质素,探究其对两种有机污染物的催化效率。有机污染物的光催化降解速率计算公式为:
-ln(Ct/C0)= -ln(At/A0)=kt
(2)
式中:A0为初始吸光度;At为t时刻吸光度;C0为污染物初始浓度,mmol/L;Ct为t时刻污染物浓度,mmol/L;t为反应时间,min;k为反应速率常数,min-1。
2 结果与分析
2.1 结构表征
2.1.1 木质素的结构表征
酶解木质素纯化前,木质素的Mn为1 867 g/mol,Mw为2 744 g/mol,PD为1.47;纯化后,木质素的分子量降低,Mn降低至855 g/mol,Mw降低至1 243 g/mol,PD约为1.45,无明显变化。纯化前,分子量大的木质素在丙酮中溶解度低是导致纯化木质素分子量降低的主要原因。纯化木质素的31P-NMR 对应主要特征峰的归属及定量分析如表1所示,纯化木质素中紫丁香基酚羟基含量为1.48 mmol/g,愈创木基酚羟基含量为2.65 mmol/g,对羟苯基酚羟基含量为1.52 mmol/g,脂肪族羟基含量为4.37 mmol/g,羧基含量为2.94 mmol/g。31P-NMR分析结果表明:纯化木质素含有丰富的还原性基团(羟基),能够将Au(Ⅲ)还原成Au(0)。

表1 由31P-NMR分析得到的木质素羟基含量Table 1 The hydroxyl contents of lignin determined by 31P-NMR
2.1.2 Au NPs的结构表征
Au NPs/木质素的TEM图见图1a,在模拟太阳光照射条件下,酶解木质素作还原剂和稳定剂,能够获得尺寸较均一且分散性良好的Au NPs/木质素。

a) TEM; b) 高分辨-TEM; c) 木质素和Au NPs/木质素的XPS光谱; d) Au 4f的高分辨率XPS光谱。图1 Au NPs的结构表征图Fig. 1 Structural characterization diagrams of Ag NPs
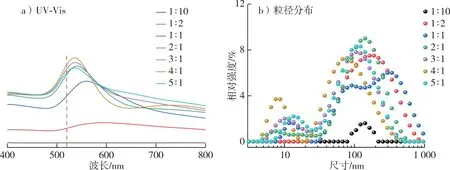
图2 不同HAuCl4和木质素溶液体积比下Au NPs紫外-可见光谱图和粒径分布Fig. 2 UV-vis spectra and particle size distribution of Au NPs prepared with different volume ratios of HAuCl4/lignin solutions
Au NPs的高分辨图像电子衍射图像(SAED)见图1b,经测量分析可知Au NPs的晶格条纹间距为0.235 nm,与文献[14]报道的Au NPs(111)晶面的晶格间距一致,Au NPs的SAED中(111)、(002)、(022)、(222)、(024)平面的出现证明了生成的Au NPs是多晶结构。采用XPS对Au NPs以及木质素的元素组成和表面化学性质进行了表征,其XPS全谱的测试结果见图1c,相较于木质素位于286.8 eV的C1s、400.2 eV的N1s和532.17 eV的O1s,Au NPs出现了新的Au 4f信号峰分别对应于Au 的4f5/2和4f7/2位于87.5和83.8 eV的特征峰(图1d),综上可知,本研究已经成功制备了Au NPs。
2.2 Au NPs/木质素制备工艺优化
2.2.1 溶液体积比
研究表明,Au、Ag、Pt等贵金属纳米粒子在紫外可见光波段展现出很强的光谱吸收,从而发生局域表面等离子体共振(LSPR)的现象,Au NPs的紫外吸收峰λ=520 nm。因此,HAuCl4溶液/木质素溶液反应后出现的UV特征吸收峰,表明溶液中Au3+被木质素溶液还原形成Au单质。此外,特征吸收峰的变化可以反映表面等离子体共振(SPR)的频率变换,SPR发生蓝移表明金属纳米粒子的粒径减小,SPR发生红移表明金属纳米粒子的粒径增大[15-16]。当HAuCl4溶液与木质素溶液体积比为1∶1时,在520 nm处出现了比较明显的特征峰(图2a),表明Au NPs的生成,并且随着溶液体积比的增大,520 nm处的吸收峰逐渐变得尖锐并出现蓝移,说明Au NPs的粒径随反应体系中Au3+增多而逐渐减小,并且当HAuCl4溶液与木质素溶液体积比为4∶1时,特征峰最尖锐且蓝移最大(图2a)。通过纳米粒度分析仪获得了Au NPs粒径分布(图2b),其中HAuCl4溶液与木质素溶液体积比在不断增大时,Au NPs平均粒径出现了先减小后增大的趋势。当两溶液体积比为4∶1时,此时Au NPs的平均粒径最小,为33.54 nm,与UV-Vis分析结果一致。这可能是因为反应体系中Au3+的增多致使Au NPs的产率随之增加,直至HAuCl4溶液与木质素溶液体积比为4∶1,体系中的Au NPs开始发生聚集,不再趋于稳定。因此,利用模拟太阳光催化制备Au NPs时,HAuCl4溶液与木质素溶液体积比为4∶1时获得的Au NPs平均粒径最小。
2.2.2 光照时间
在不同光照时间下进行Au NPs的还原实验,结果见图3。如图3a所示,当模拟太阳光照射10 min 时,在反应体系中观察到SPR峰的出现,表明有Au NPs生成。随光照时间的增加,SPR强度逐渐增强,当光照时间达30 min时,SPR峰的强度不再变化,表明Au NPs在该溶液体系中完成了还原;继续进行光照60 min时,SPR峰位较30 min出现蓝移,随后SPR峰又开始红移,表明生成的Au NPs粒径先减小后增大,因此光照催化时间为60 min时Au NPs粒径达到最小。这一现象出现的原因可能是随着反应时间的增加,体系中不断产生Au NPs而后发生聚集。利用纳米粒度仪测量不同光照时间下的Au NPs粒径分布,如图3b所示,随着光照时间的增加,Au NPs的粒径在光照60 min时最小(32.68 nm),与SPR峰测量结果一致。因此,模拟太阳光催化制备Au NPs最佳光照时间为60 min。
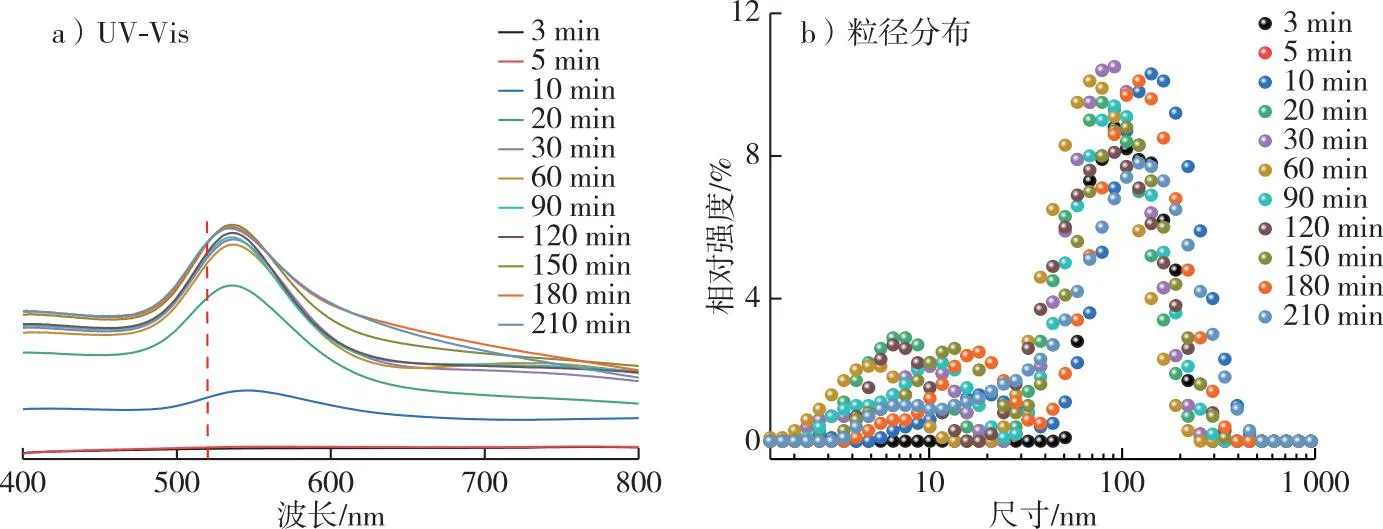
图3 不同光照时间下Au NPs紫外-可见光谱图和粒径分布Fig. 3 UV-vis spectra and particle size distribution of Au NPs prepared with different illumination times
光照30,60和90 min制得的Au NPs的TEM形貌图见图4。结果表明,光照时间对Au NPs的形貌及尺寸影响较小。当光照时间为30 min时,Au NPs的平均粒径为41.63 nm;当光照时间为60 min时,Au NPs的平均粒径为32.68 nm;当光照时间为90 min时,Au NPs的平均粒径为39.98 nm。

a) 30 min; b) 60 min; c) 90 min。图4 不同光照时间生成的Au NPsFig. 4 Morphology of Au NPs prepared with different light irradiation times
2.2.3 HAuCl4浓度
当HAuCl4溶液与木质素溶液体积比和光照时间固定时,反应体系中适当的Au3+浓度可以使木质素迅速将Au3+还原成Au NPs[10]。不同HAuCl4浓度下木质素还原Au NPs的SPR谱图见图5。如图5a所示,Au NPs的SPR峰随HAuCl4浓度增大而逐渐增强,说明溶液中生成的Au NPs不断增多。当HAuCl4浓度为1.00 mmol/L时,Au NPs在520 nm处的SPR尖锐且峰值高,说明此时Au NPs粒径分布窄且产量较高;当HAuCl4浓度增大至1.50 mmol/L时,SPR峰值到达最大,继续增大浓度,SPR峰值开始逐渐减弱,半峰宽变大,峰位红移,此时生成的Au NPs因体系中Au3+的饱和而发生团聚导致粒径增大,Au核浓度也随之降低。不同HAuCl4浓度下生成的Au NPs粒径分布图见图5b。由图5b可知,随着HAuCl4浓度的增加,Au NPs粒径表现出逐渐减小的趋势。直至HAuCl4浓度增大至1.00 mmol/L时,Au NPs的平均粒径最小,为32.41 nm;此后随着HAuCl4浓度的继续增加,生成的Au NPs无法均匀分散在体系中而发生团聚,粒径逐渐增大,当HAuCl4浓度为3.00 mmol/L时,Au NPs的平均粒径可达703.8 nm。
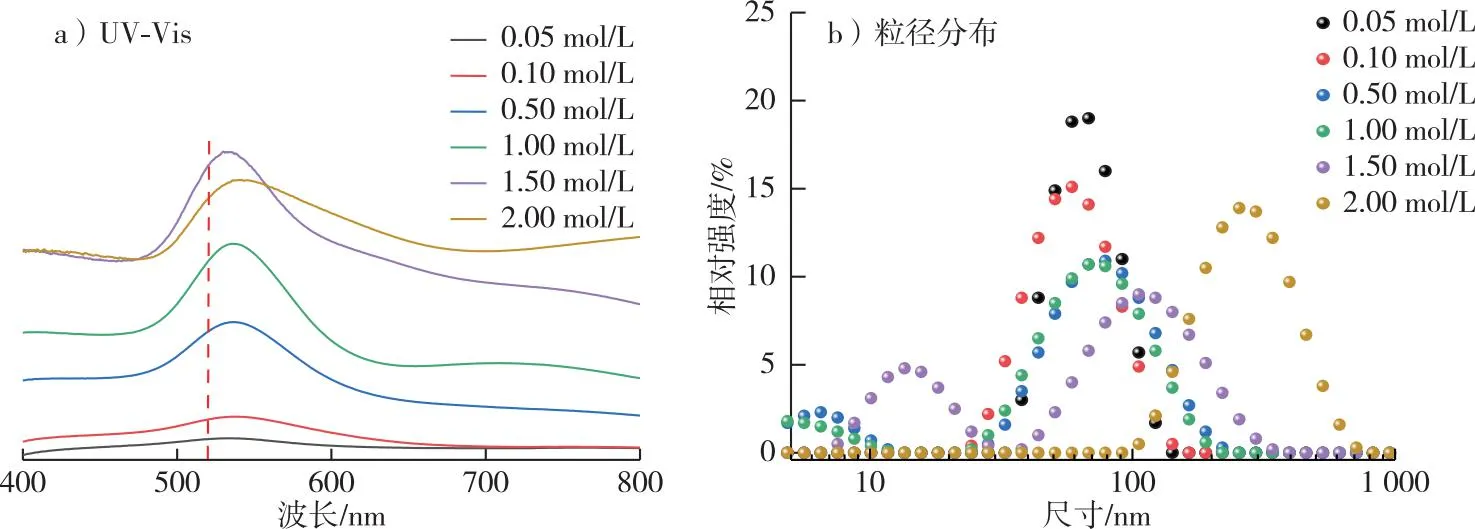
图5 不同HAuCl4浓度下Au NPs紫外-可见光谱图和粒径分布Fig. 5 UV-vis spectra and particle size distribution of Au NPs prepared with different concentrations of Au3+
在光照条件下,控制不同浓度的HAuCl4溶液得到Au NPs的形貌如图6所示。当HAuCl4浓度较低时,Au3+被还原成Au核后,由于Au3+不足,导致Au停止生长,稳定分散在溶液中(图6a和b);随着Au3+浓度的增加,被还原的Au核也越来越多,Au3+开始附着在Au核表面上生长从而出现了不同形貌(图6c);当HAuCl4浓度为1.00 mmol/L时,生成的Au NPs浓度较0.50 mmol/L时更高,尺寸更均一(如图6c和d);当HAuCl4浓度继续增加至1.50 mmol/L时,Au NPs开始出现不同程度的团聚(图6e),继续增加HAuCl4浓度,团聚现象加剧(图6f)。这与图5a中Au3+高于1.50 mmol/L时,Au NPs 的SPR峰强度降低,并伴随着红移现象的出现一致。因此,利用木质素在模拟太阳光催化下制备Au NPs最佳的HAuCl4溶液浓度为1.00 mmol/L。

a) 0.05 mmol/L; b) 0.10 mmol/L; c) 0.50 mmol/L; d) 1.00 mmol/L; e) 1.50 mmol/L; f) 2.00 mmol/L。图6 不同浓度HAuCl4制得Au NPs的TEM图Fig. 6 Morphology of Au NPs prepared with different concentrations of Au3+
2.3 Au NPs/木质素的催化性能
2.3.1 Au NPs/木质素催化还原模型污染物
室温下,以MB和4-NP作为有机污染物模型探究Au NPs/木质素分散体系的催化活性。利用UV-Vis分别测定MB和4-NP在664和400 nm处的紫外吸收峰强度变化[14,17]。在3 mL MB(0.05 mmol/L)和NaBH4(0.05 mmol/L)溶液中加入15 μL Au NPs/木质素分散液(1 mmol/L),MB在664 nm处的特征吸收峰强度随时间的增加而逐渐减弱,并在5 min后趋于平缓(图7a)。MB的催化还原率在5 min内达97%,MB几乎完全被还原;同样地,加入20 μL Au NPs/木质素分散液(1 mmol/L)时催化还原率在3.3 min内达95%(图7b);而当反应体系中只有MB 和NaBH4存在时,即使均匀混合12 min,MB在664 nm处的吸收峰也几乎不发生变化,这说明没有Au NPs存在时,MB未发生还原反应(图7c)。在3 mL 4-NP(0.05 mmol/L)和NaBH4(0.05 mmol/L)溶液中加入50 μL Au NPs/木质素分散液(1 mmol/L),随着反应时间的增加,4-NP在400 nm处的特征吸收峰强度逐渐减弱(图7d)。同时,300 nm处新出现的紫外吸收峰,表明4-NP被逐渐催化还原成4-氨基苯酚(4-AP)。反应进行到7.3 min时,Au NPs对4-NP的催化还原率达82%;Au NPs/木质素分散液(1 mmol/L)加入量为80 μL时,反应5.7 min后Au NPs催化还原4-NP高达83%(图7e),Au NPs/木质素分散液加入量的增加使其对4-NP的催化还原时间明显缩短;当反应体系中只有4-NP和NaBH4时,即使混合反应12 min, 4-NP在400 nm处的紫外吸收峰也基本不变(图7f), 这说明体系中无Au NPs/木质素存在时,4-NP未发生催化还原。实验结果表明,Au NPs/木质素分散体系对MB和4-NP均有良好的催化还原作用。
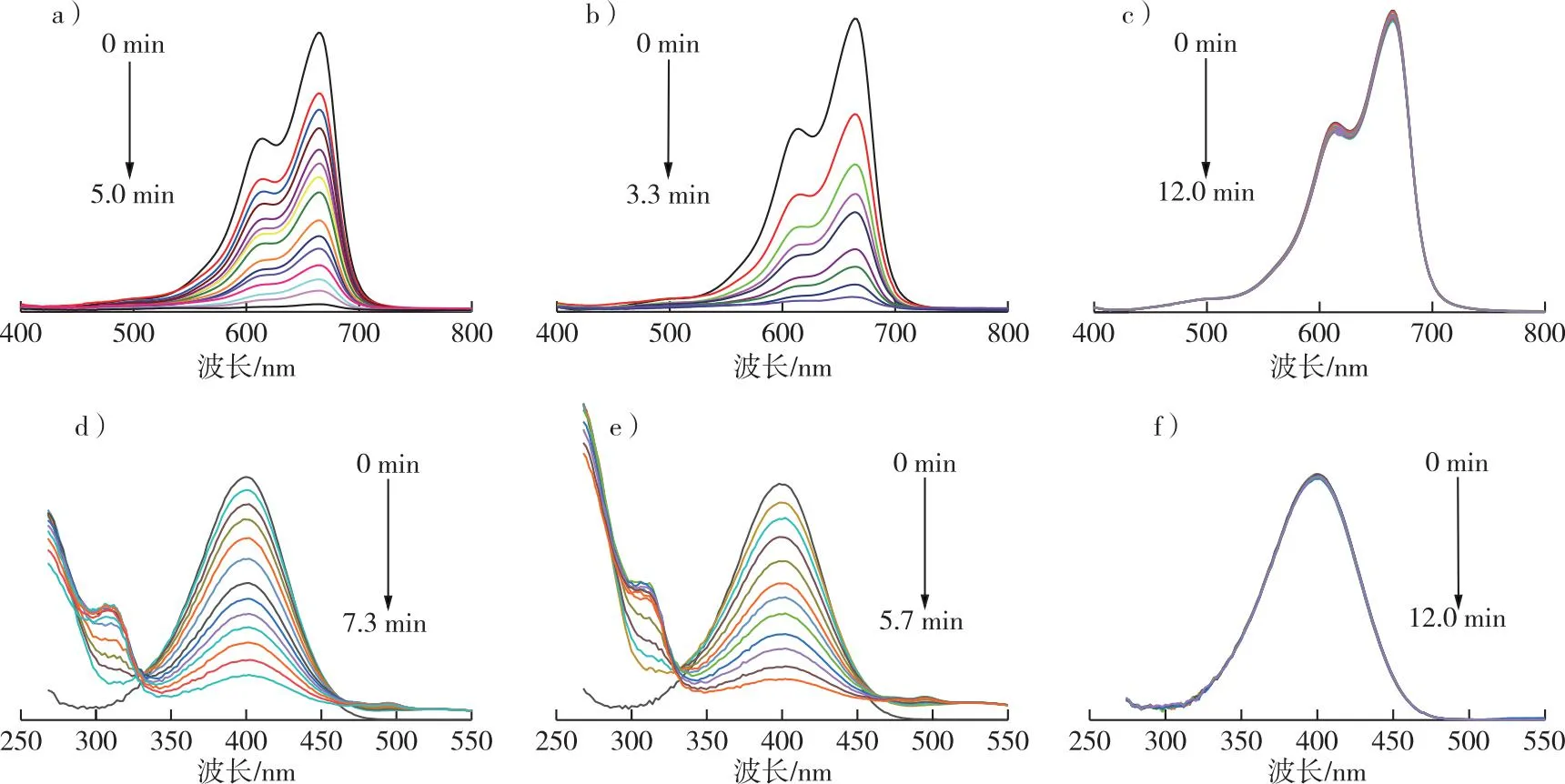
a) 15 μL Au NPs (1 mmol/L) (MB+NaBH4); b) 20 μL Au NPs (1 mmol/L) (MB+NaBH4); c) 无Au NPs (MB+NaBH4); d) 50 μL Au NPs (1 mmol/L) (4-NP+NaBH4); e) 80 μL Au NPs (1 mmol/L) (4-NP+NaBH4); f) 无 Au NPs (4-NP+NaBH4)。图7 MB和4-NP在664和400 nm处的紫外吸收峰强度变化Fig. 7 UV-vis spectra of MB and 4-NP solutions added with different Au NPs solutions
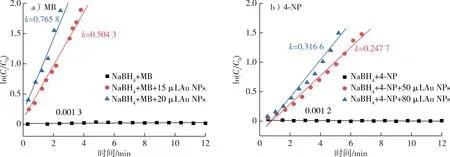
图8 Au NPs催化NaBH4催化还原准一级动力学Fig. 8 Quasi first-order kinetics of reduction of MB and 4-NP by NaBH4 catalyzed with Au NPs
2.3.2 Au NPs/木质素催化还原动力学分析
为了进一步探究Au NPs/木质素对MB和4-NP的催化还原活性,通过准一级动力学计算反应速率常数k值评估反应的催化速率,k值越大,催化反应速率越快、反应时间越短,说明Au NPs/木质素的催化性能越好。根据ln(Ct/C0)与反应时间之间的线性关系计算反应速率常数k,结果如图8所示。20 μL Au NPs对MB和NaBH4的催化反应速率k值为0.765 8 min-1,80 μL Au NPs对4-NP和NaBH4的催化反应速率k值为0.316 6 min-1,说明催化剂Au NPs/木质素对MB、4-NP均有较好的催化还原活性,且反应动力学常数随Au NPs/木质素加入量的增加而增加。基于以上实验结论,提出Au NPs/木质素在该条件下对于MB的催化反应机理:NaBH4在水溶液中首先电离出 Na+离子和 BH4-离子,BH4-离子提供电子诱导MB的还原;在不存在催化剂的条件下,该反应的发生需要越过一个较高的能量势垒,因而该反应自发过程十分缓慢。然而,当体系中引入Au NPs后,Au NPs 则充当了两者间电子转移的“桥梁”,MB分子和 BH4-离子与 Au NPs接触时,MB分子从Au NPs催化剂表面获得电子,随之立即被还原和解吸,为后续的MB分子释放一个新的还原位点,并重复上述过程以确保还原反应的连续性。4-NP的还原机理同上,4-NP分子从Au NPs催化剂表面获得电子后被立即催化还原,最终达到较高催化还原水平。
3 结 论
利用一种简单的光催化木质素还原Au(Ⅲ)的方式制备了具有优异催化还原性能的Au NPs/木质素,深入探究了Au3+浓度、木质素质量分数、光照时间等不同变量对Au NPs尺寸和形貌的影响,以MB和4-NP为模拟污染物,探究Au NPs/木质素的催化性能,揭示其光催化机理,具体结论如下:
1)纯化木质素溶液作为Au3+的还原剂和稳定剂,在光催化条件下,通过调控HAuCl4溶液/木质素溶液体积比、光照时间以及HAuCl4浓度可以制得粒径可控的Au NPs。
2)制备Au NPs的最佳工艺条件为:HAuCl4与木质素体积比4∶1,光照时间60 min,HAuCl4浓度1.00 mmol/L,其平均粒径为32.41 nm。
3)催化降解动力学分析证明Au NPs/木质素对MB(3.3 min,95%)和4-NP(5.7 min,83%)均有较好的催化还原活性。
