Characteristics of cross transmission of gut fungal pathogens between wintering Hooded Cranes and sympatric Domestic Geese
2024-01-22YunnuoWuXioyuFnJieYuTinciLiuRongCuiXingjiXing
Yunnuo Wu, Xioyu Fn, Jie Yu, Tinci Liu, Rong Cui, Xingji Xing,c,*
aSchool of Resources and Environmental Engineering,Anhui University,Hefei,230601,China
bAnhui Province Key Laboratory of Wetland Ecosystem Protection and Restoration,Hefei,230601,China
cInternational Collaborative Research Center for Huangshan Biodiversity and Tibetan Macaque Behavioral Ecology,Hefei,230601,China
Keywords:Cross transmission Fungi Gut pathogen Migratory bird Poultry
ABSTRACTMigratory birds travel long distance and link various pathogens.Due to habitat degradation, wintering waterfowls forage together with poultry, increasing the risk of pathogen transmission between hosts.We investigated the fungal communities between wintering Hooded Cranes and Domestic Geese by high-throughput sequencing,and inferred the potential gut pathogens for both hosts at different wintering stages.The fungal community compositions were significantly different between seasons and between the hosts.The negative correlation was found between fungal diversity and pathogenic diversity, with higher fungal diversity and less pathogenic diversity in early stage and less fungal diversity and higher pathogenic diversity in late stage for both hosts.The dissimilarity of pathogenic community compositions decreased from early to late stage, with the quantity of overlapping potential pathogenic OTUs increasing along wintering periods between the two hosts, demonstrating the existence of pathogen cross transmission between the two hosts.Furthermore, the transmission pathway of avian pathogens was mainly from Hooded Cranes to Domestic Geese while the transmission direction of human pathogens was primarily from Domestic Geese to wild cranes.Based on above results, we argued that pathogen cross transmission and underlying outbreak risk of disease should be closely monitored in migratory birds and domestic poultry.
1.Introduction
The animal gut contains complex microbial communities which form a network system (Bletz et al., 2016; Lozano et al., 2019).Certain interaction has been demonstrated between the host and their gut microbial community (Wang et al., 2018).The hosts can affect their gut microbial community by shifting community assembly process (Wong et al., 2015).Gut microbiota provide necessary energy for their hosts as probiotics, and also harm the health of hosts as pathogens (Hager et al.,2019).
The avian guts act as important sites for microbial colonization and host-microbe interactions (Cresci and Bawden, 2015; Bodawatta et al.,2022).Correlative studies have demonstrated the existence of intricate network in gut microbiota of birds, and the bird gut microbiota is highly dynamic (Paul et al., 2021; Robinson et al., 2022; Wang et al., 2022).Migratory birds have the cross-region migration habit, exhibiting strong geographical diffusion capabilities and extensive distribution characteristics.The periodic migration of migratory birds contacts with different habitats and intermediary media, making the sources of pathogens more diverse and complex (Huang et al., 2014).Thus,migratory birds may link various pathogens and spread their pathogens across regions.
Studies have found that many outbreaks of poultry diseases were closely connected with migratory bird migration and often synchronized the arrival time of migratory birds (Humphreys et al., 2020, 2021).Around the wintering grounds, a large number of poultry often forage together with waterbirds in the same area.The pathogens carried by wild birds may spread to poultry through shared niche (Grond et al.,2014; Fu et al., 2020).Similarly, pathogens in poultry can also be transmitted to migratory birds in the same manner, forming a continuously circulating two-way transmission pattern (Pantin-Jackwood et al.,2016; Lewis et al., 2021).In addition, avian pathogens can infect humans through poultry vectors, which can pose a serious threat to human health (Mora et al., 2013; Mercan et al., 2021).
The Shengjin Lake is an important gathering area for migratory waterfowls in the middle and lower reaches of the Yangtze River.In recent years, the suitable habitat for migratory birds has been continuously lost due to intensified human activities and lake degradation,which forces migratory birds to forage in paddy fields and grasslands,exacerbating the overlapping of ecological niches with poultry (Yang et al., 2015).This may increase the risk of pathogen cross transmission between migratory birds and poultry.The Hooded Crane (Grus monacha)is a national level I key protected wild animal in China, listed as Vulnerable (VU) species on the IUCN Red Species List (Huang et al.,2014).According to our recent surveys, about 300–350 Hooded Cranes arrived at Shengjin Lake to overwinter each year, and in this research it was 320 cranes.These Hooded Cranes utilized both natural wetlands and neighboring paddy fields for foraging.In wintering periods, the Hooded Cranes might spend more time in paddy fields to forage.When Hooded Cranes foraged in paddy fields, they shared niche with Domestic Geese (Anser anser domesticus).Current research focused on pathogenic bacteria in migratory birds and poultry (Xiang et al., 2023), but little is known about the characteristics of cross transmission of fungal pathogens between them.
In this research, we use the high-throughput sequencing method to analyze intestinal fungal community in wild Hooded Cranes and Domestic Geese in the wintering periods.The purposes of this research were: 1) to clarify the seasonal dynamic patterns of gut fungal community between wintering Hooded Cranes and Domestic Geese, and infer the community structure of intestinal pathogens between the two hosts; and 2) to assess the degree and characteristics of cross transmission of various types of pathogens between hosts under seasonal dynamic patterns.
2.Materials and methods
2.1.Sample collection and species determination
The Shengjin lake served as a vital wintering area for long-distance migratory wild birds (Xiang et al., 2019).The fecal samples were collected at the Shengjin Lake in two stages: the early stage from November 11thto November 13th, 2018; the late stage from February 19thto February 21th, 2019.A total of 80 samples of Hooded Cranes and Domestic Geese were collected, with 40 for each species (i.e., 20 samples each from early and late wintering stages, respectively).The DNA of samples were extracted by the Qiagen DNA Stool Mini kit.After DNA extraction, bird species determination was performed by amplification ofCOIgene for sequencing.Detailed information for site selection,sample collection, DNA extraction and species determination was shown in the Supplementary data.
2.2.Bioinformatics
The primer set ITS1/ITS2 (ITS1: 5′-CTTGGTCATTTAGAGGAAGTAA-3′; ITS2: 5′-GCTGCGTTCTTCATCGATGC-3′) was used to conduct the PCR for ITS gene sequencing.The detail information of PCR and amplicon library preparation could be found in the Appendix.Raw fungal data were processed by QIIME 1 (QIIME 1.9; Caporaso et al.,2010).High-quality sequences (average mass fraction more than 30 and length >250 bp) were clustered into Operational Taxonomic Units(OTUs) by the UCLUST (Edgar, 2010).The OTUs of chimeras were filtered by the ‘usearch’ method.A representative sequence was picked for each OTU and identified by the ribosomal database project Classifier(Caporaso et al., 2010).For similarly sparse samples, we selected 29,000 sequences at random in each sample to generate a new subset of sequences for further analysis.
2.3.The determination of pathogenic species
All detected fungal species were retrieved as keywords in Web of Science to identify the pathogenic species.Pathogenic fungal species that have been shown in two or more references as potential pathogens and could lead to associated diseases in various animals and/or human were selected for further analysis.
2.4.Statistical analysis
The one-way ANOVA and two-way ANOVA tests were conducted to evaluate the differences of alpha-diversity for Hooded Crane and Domestic Geese at early and late stages.Fungal community composition was shown by principal component analysis (PCA).The fungal community assembly was evaluated by Sloan neutral model (Luan et al.,2020).Co-occurrence network was performed to infer the interaction within gut fungi of the two hosts.The Cytoscape software was used to depict the overlapping of pathogenic OTUs.The pathogenic diversity and relative abundance were estimated by two-way ANOVA andt-test.The composition of pathogenic community was analyzed by non-metric multidimensional scaling (NMDS, Bennett et al., 2008) and similarity analysis (ANOSIM, Keenum et al., 2021).The zetadiv R package was used to calculate zeta diversity to estimate higher-order compositional similarity among multiple samples.The zeta diversity of this study represented the similarity of community composition among individuals of a population.When the curve declines sharply, it means the similarity of community composition rapidly decreased along the increasing number of samples (Hui and McGeoch, 2014; Latombe et al., 2018;McGeoch et al., 2019).The detail information can be found in Appendix.
3.Results
3.1.The gut fungal communities under seasonal dynamic patterns
A total of 4,748,552 high quality sequences with 4465 fungal OTUs were detected in all samples, ranging from 29,580 to 72,622 sequences and 40 to 748 OTUs per sample, respectively.The two-way ANOVA showed that wintering stage rather than host species significantly affected gut fungal alpha-diversity (Appendix Table S1).Gut fungal Chao1 index and OTU richness were significantly higher at the early stage compared to the late stage for both hosts (Fig.1A and B).However,in each stag, there was little difference of gut fungal diversity between Hooded Cranes and Domestic Geese (Fig.1A).Significant differences of fungal community compositions were found between different wintering stages and between host species (Fig.1C).However, the differences in gut fungal community composition decreased significantly from early stage (r= 0.709***) to late stage (r= 0.100**) between Hooded Cranes and Domestic Geese (Fig.1C).The fungal compositional similarity decreased sharply in late stage than early stage in guts of Domestic Geese (Fig.1D).The Sloan neutral community model was used to evaluate the avian gut fungal community assembly (Appendix Fig.S1).The fits and estimated migration rates of neutral model for both Hooded Cranes and Domestic Geese tended to decline, with the relative importance of neutral process decreasing while niche process increasing along the wintering periods.
Co-occurrence network was used to analyze the interaction within gut fungal taxa of wintering cranes and Domestic Geese.The result showed that most of the connections in gut networks of the two hosts were related to Ascomycota (Appendix Fig.S2).In both two stages,Hooded Cranes possessed higher average degree, density, clustering coefficient and the lower average path length relative to Domestic Geese(Appendix Table S2).To assess network stability through natural connectivity, with the higher network stability in late stage than that in early stage between the two hosts (Appendix Fig.S3).
3.2.The potential pathogens under seasonal dynamic patterns
A total of 404,187 potential animal pathogenic sequences and 239 pathogenic OTUs were detected across all samples.These pathogenic OTUs belonged to 38 pathogenic species (Appendix Table S3).The quantity of shared gut pathogenic OTUs were 49 between Hooded Cranes and Domestic Geese at the early stage, and increased to 149 at the late stage (Fig.2A and B; Appendix Table S4).The unique pathogenic OTUs of Hooded Cranes and Domestic Geese showed a large increase from early to late stages (Fig.2C and D).Among these pathogenic sequences, we found a total of 304,015 potential avian pathogenic sequences with 117 OTUs, and a total of 49,743 potential human pathogenic sequences with 66 OTUs.The quantity of 18 shared avian pathogenic OTUs were detected between Hooded Cranes and Domestic Geese at the early stage, and increasing to 82 shared OTUs at the late stage (Fig.2E and F; Appendix Table S5).The unique avian pathogenic OTUs of Hooded Cranes and Domestic Geese increased from early to late stage (Fig.2G and H).The number of shared human pathogenic OTUs between Hooded Cranes and Domestic Geese increased from seven at early stage to 32 at the late stage (Fig.2I and J; Appendix Table S6).The unique pathogenic OTUs of both Hooded Cranes and Domestic Geese increased from early to late stage (Fig.2K and L).
The two-way ANOVA showed that wintering stage and host species significantly affected gut fungal pathogen (Appendix Table S7).The pathogenic diversity showed significantly higher in late stage relative to early stage in both hosts (Appendix Fig.S4).For Hooded Crane, the relative abundance of avian pathogen showed little difference between two stages while the relative abundance of human pathogen showed significantly higher in late stage (Appendix Figs.S4C and K).For Domestic Geese, the relative abundance of pathogen showed significantly higher in late stage than early stage (Appendix Figs.S4D and H, L).In early stage, Hooded Cranes had higher pathogenic diversity and relative abundance relative to Domestic Geese while in late stage they showed little difference (Fig.3A-D).The pathogenic community compositions showed significant differences between the two stages and between two host species (Fig.3E).The non-similarity value (r) of pathogenic community composition between Hooded Cranes and Domestic Geese significantly decreased from 0.882*** (rvalue) in early stage to 0.145**(rvalue) in late stage (Fig.3E).
In early stage, Hooded Cranes had higher avian pathogenic diversity and relative abundance relative to Domestic Geese while they showed little difference in late stage (Fig.3F-I).The non-similarity value of avian pathogenic community composition between Hooded Cranes and Domestic Geese markedly decreased from 0.889*** (rvalue) in early stage to 0.062* (rvalue) in late stage (Fig.3J).In early stage, Hooded Cranes harbored less diversity (Fig.3K) and relative abundance(Fig.3M) of human pathogen relative to Domestic Geese.The human pathogenic diversity showed little difference between Hooded Cranes and Domestic Geese in late stage (Fig.3L).The Hooded Cranes had higher relative abundance of human pathogen relative to Domestic Geese in late stage (Fig.3N).The non-similarity value of human pathogenic community composition between Hooded Cranes and Domestic Geese decreased from 0.388*** (rvalue) in early stage to 0.299*** (rvalue) in late stage (Fig.3O).
The result of Zeta diversity showed larger compositional similarity of pathogen in late stage than early stage in guts of Hooded Cranes(Fig.4A).The compositional similarity of pathogen decreased sharply in late stage than early stage with a cross in order 16 in guts of Domestic Geese (Fig.4A).The larger compositional similarity of avian pathogen was found in late stage than early stage in guts of Hooded Cranes.The compositional similarity of avian pathogen decreased sharply in late stage than early stage with a cross in order 14 in guts of Domestic Geese(Fig.4B).The larger compositional similarity of human pathogen was found in late stage than early stage in both guts of two hosts (Fig.4C).
4.Discussion

Fig.3.The diversity, relative abundance and community composition of various gut pathogens in early and late stages between Hooded Cranes and Domestic Geese.Bars in represent mean values; error bars indicate standard deviation.Letters indicate significant differences fromt-test (P< 0.05).Non-metric multidimensional scaling analysis (NMDS) and analysis of similarity (ANOISM) in figure E-J-O analyze the differences of gut pathogenic fungal community between the two hosts at different wintering stages.EHC: the early stage of hooded cranes; LHC: the late stage of hooded cranes; EDG: the early stage of domestic geese; LDG: the late stage of domestic geese.*: 0.01 Empirical researches have shown that host species and season variation were important factors shaping the gut microbiota (Xue et al.,2015; Wen et al., 2019).Fungal community assembly (stochastic vs.deterministic) processes could modulate microbial community structure(Zhang et al., 2022).The increasing relative importance of deterministic process could strengthen the effect of intestinal selection (Menéndez--Serra et al., 2023) to affect the fungal community composition (Zhou and Ning, 2017; Yang et al., 2021; Wu et al., 2022).The gut network displayed the more complicacy with the higher stability and average degree, and the lower average path length in Hooded Cranes relative to Domestic Geese.The higher average degree and lower average path length were conducive to better organization of metabolic pathways and information exchange within gut fungal taxa (Zhou et al., 2010).The network with higher stability could be related to greater resistance to environmental undulation and supported more versatility (Deng et al.,2012; Yang et al., 2021).Thus, Hooded Cranes might be more capable of adapting to the local environment by regulating their microbiota compared to Domestic Geese during the wintering periods. The fungal pathogens were detected, with higher pathogenic diversity in the late stage for both hosts.However, the fungal diversity was significantly higher in guts of cranes and geese in early stage relative to late stage.The results were consistent with the previous study,showing the negative correlation between pathogenic diversity and microbial diversity in guts of animals (Bajaj et al., 2018; Xiang et al.,2019; Zhang et al., 2023).In this study, the dissimilarity (r) of pathogenic community decreased from early stage to late stage, with the overlapping OTUs of potential pathogen rose from early to late stage between the two hosts.Previous studies have demonstrated that the overlapping of ecological niches might be a crucial way leading to the cross transmission of pathogens among different host species (Maher et al., 2010; Corti et al., 2022).Hooded Cranes often mixed flock with Domestic Geese to forage (i.e., shared niche) at paddy fields, which caused the pathogen cross transmission between the two species, and thus the higher unique pathogenic OTUs in late stage than that in early stage for both hosts. Fig.4.The zeta diversity in figure A-B-C is the average number of OTUs shared between a specific number of samples, calculated for each group and order from a combination of 10,000 non-overlapping samples.OTU: operational taxonomic units.EHC: the early stage of hooded cranes; LHC: the late stage of hooded cranes;EDG: the early stage of domestic geese; LDG: the late stage of domestic geese. In early stage, the diversity and relative abundance of avian pathogens were higher in Hooded Cranes than Domestic Geese.Hooded Cranes inhabited different habitats and contacted with many other birds among migratory process, which might make them be exposed to a complex and diverse range of avian pathogens.Domestic Geese had a limited range of movement and limited exposure to pathogens.Pathogens in Hooded Cranes could be easily transmitted to Domestic Geese,triggering the rise of their pathogenic diversity and relative abundance after shared the ecological niches by mixing forage of two hosts (Xiang et al., 2021).Thus, the transmission of avian pathogens was primarily from wild Hooded Cranes to Domestic Geese, which was consistent with previous study showing that the transmission of bacterial pathogens was mainly from wild birds to Domestic Geese (Xiang et al., 2023).The Zeta diversity showed that the compositional similarity of avian pathogens in Domestic Geese decreased sharply in late stage, suggesting that the spread of avian pathogens from Hooded Cranes to Domestic Geese were specialized and might occur in only a small number of Domestic Geese.This pattern might be induced by uneven contact of sampled geese with Hooded Cranes or the application of antibiotics for poultry. The human pathogens were detected in both Hooded Cranes and Domestic Geese, suggesting that wild birds and/or poultry might be important vectors for spread of human disease (Xiang et al., 2023).The Domestic Geese contacted with human beings frequently, thus they carried more human pathogens than wild birds.However, in late stage,we found significant increase of diversity and relative abundance of human pathogens in Hooded Cranes.We speculated that there might be a main directional transmission pathway of human pathogens from Domestic Geese to Hooded Cranes.These human pathogens from poultry to wild birds could be spread with long distance by avian migration, posing a risk to human health across regions (Alm et al.,2018).The zeta diversity of human pathogen in guts of Hooded Cranes was high and decreased slowly at the late stage, indicating that the human pathogens from Domestic Geese could be widely transmitted within Hooded Crane population, possibly because of wild birds without application of antibiotics. This study investigated the seasonal dynamic patterns of the fungal community and pathogenic community in Hooded Cranes and Domestic Geese during the wintering period.Our results demonstrated that overlapping foraging niches might have contributed to the cross transmission of fungal pathogens between Hooded Cranes and Domestic Geese.Avian pathogens might have a main directional transmission pathway from wild Hooded Cranes to Domestic Geese, while human pathogens harbored a major transmission pathway from Domestic Geese to Hooded Cranes.We argued that the transmission pathway of pathogens between migratory birds and domestic poultry and underlying outbreak risks of diseases should be closely monitored.As Hooded Crane is an IUCN Vulnerable species, the specific natural reserves should be established for them to avoid overlapping ecological niches with domestic animals.In addition, the poultry industry should be reduced around the reserve to offer a better living environment for wild Hooded Cranes. Funding This study was financially supported by the “National Natural Science Foundation of China” (Grant No.31801989) and the Outstanding Youth Research Project of Anhui Province for Xingjia Xiang(2022AH030015). CRediT authorship contribution statement Yuannuo Wu:Conceptualization, Formal analysis, Methodology,Writing – original draft, Writing – review & editing.Xiaoyu Fan:Data curation, Formal analysis, Methodology.Jie Yu:Data curation.Tianci Liu:Data curation.Rong Cui:Data curation.Xingjia Xiang:Conceptualization, Formal analysis, Methodology, Writing – original draft,Writing – review & editing. Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Acknowledgments We thank Miss Rong Fu from Anhui University for assistance in sample collection. Appendix.Supplementary data The raw data were submitted to the Sequence Read Archive (SRA) of NCBI under the accession number SAMN35669197-SAMN35669200.Supplementary data to this article can be found online at https://doi.org/10.1016/j.avrs.2023.100142.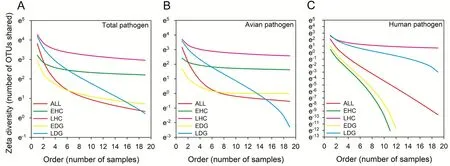
5.Conclusions
杂志排行

Avian Research的其它文章
- Selecting the best: Interspecific and age-related diet differences among sympatric steppe passerines
- Morphology and morphometry of two hybridizing buntings at their hybrid zone in northern Iran reveal intermediate and transgressive morphotypes
- Quiet in the nest: The nest environment attenuates song in a grassland songbird
- Fecal DNA metabarcoding reveals the dietary composition of wintering Red-crowned Cranes (Grus japonensis)
- Short-term night lighting disrupts lipid and glucose metabolism in Zebra Finches: Implication for urban stopover birds
- Variaiton in the composition of small molecule compounds in the egg yolks of Asian Short-toed Larks between early and late broods