基于D-(-)-/L-(+)-对羟基苯甘氨酸的两对手性钴配合物的合成、晶体结构和电化学识别
2024-01-20曹梦茹姜国远李宏利李思浓宋会花
曹梦茹 姜国远 李宏利 李思浓 宋会花
(河北师范大学化学与材料科学学院,河北省有机功能分子重点实验室,石家庄 050024)
0 Introduction
Chirality is a universal aspect of the chemical sciences[1-2].Recognition of chiral amino acids plays a pivotal part in chemical biology and pharmacology, since different enantiomeric amino acids may make quite a difference in biological behavior and pharmacological activity[3-6].L-histidine(L-His),as a semi-essential amino acid,can alleviate some physiological illnesses such as heart disease, anemia, and rheumatoid arthritis,whileD-histidine (D-His) has no nutritional value to the human body.It is often used in scientific research to study the structure-function analysis of enzymes[7-11].Thus, exploiting effective and practical techniques for chiral analysis of His enantiomers in the biochemical analysis is meaningful.Conventional detection technologies for His enantiomers mainly include high-performance liquid chromatography, fluorescence,etc[12-13].However, these methods require high concentrations of analytes, sophisticated operation, and relatively expensive instrumentation, and HPLC is also typically timeconsuming[14].In this context, the highly selective, lowcost,fast-speed,real-time,and online operation electrochemical sensors have been evaluated as a promising chiral analysis technique[15-18].
The key to electrochemical chiral sensing is the material used to prepare the sensor′s electroactive surface.The chiral coordination compounds (CCPs) are an intriguing class of crystalline materials formed usually by the self-assembly of metal ions and chiral polydentate ligands[19-21].These materials are highly promising for electrochemical chiral sensors owing to the ultrahigh surface area, precise network structures, and their exposed active sites of ligand groups and metal ions that provide selective recognition and boost the charge transports[22].So far, electrochemical methods based on CCPs have had limited reports for the identification of chiral isomers because few applications were developed based on the current difference in voltammetry.For instance, Rufina A.Zilberg et al.synthesized mixed chelate compounds [M(S-Ala)2(H2O)n][M(S-Phe)2(H2O)n] (M=Cu(Ⅱ), Zn(Ⅱ);n=0-1), which can be used to analyze Nap and Prp enantiomers[23].Therefore, CCPs are a promising electrode modification material for electrochemical chiral recognition.
Among the synthetic methods of chiral coordination compounds, using the chiral ligands is the most direct and effective method[24-31].Amino acids and their derivatives have both amino and carboxyl functional groups, which exhibit excellent coordination capabilities.The carbonyl oxygen atoms, hydroxyl oxygen atoms, and nitrogen atoms can form hydrogen bonds and further form supramolecular structures.Chiral amino acid derivatives have also been confirmed to have some contributions in the research of recognition performance[32].D-(-)-/L-(+)-4-Hydroxyphenylglycine (D-/LHhpg) as amino acid derivative chiral ligands have natural advantages due to their being cheap, easily available, non-toxic, and harmless.We adopt a mixedligand strategy that selectsD-Hhpg andL-Hhpg as chiral ligands with N-donor ligands and transition Co (Ⅱ)metal ions to construct chiral coordination compounds.
With this background information, herein two pairs of chiral coordination compounds {[Co(D-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n(1-D), {[Co(L-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n(1-L),[Co(D-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-D),and [Co(L-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-L), where 4,4′-bipy=4,4′-bipyridine, 5,5′-BM-2,2′-bipy=5,5′-dimethyl-2,2′-bipyridine, have been successfully synthesized and characterized by elemental analysis, X-ray photoelectron spectroscopy(XPS), infrared spectroscopy, thermogravimetric analysis(TGA),powder X-ray diffraction(PXRD),and singlecrystal X-ray diffraction.The effects of N-donor ligands on the structures of the compounds have been discussed in detail.Their electrochemical behaviours and electrochemical recognition were further studied via cyclic voltammetry (CV) and differential pulse voltammetry (DPV).Notably, compound 1-D behaved high electroconducting and distinctive oxidation signals for each enantiomer ofD-/L-His.Not only is the sensor able to discriminate enantioselectively, but also it allows for the rapid quantitative determination of enantiomeric excess in the His mixture.Compared with other materials requiring multistep syntheses, it is an extraordinary and meaningful work to use cheap and easy synthesized crystals for enantiomeric identification.
1 Experimental
1.1 Material and measurements
All chemicals and solvents in the syntheses were purchased from commercial sources and were used as received without further purification.IR spectra were collected on an FTIR-8900 spectrometer within the 4 000-400 cm-1region (KBr pellets).TGA experiments were carried out on a simultaneous STA 449F3/TENSOR 27 thermal analyzer under a static N2atmosphere with a heating rate of 10 ℃·min-1from room temperature to 800 ℃.Elemental analyses were performed on an Elemental Vario EL elemental analyzer.XPS spectrum was recorded on a Thermascientific ESCALAB QXi instrument with AlKαX-rays as the excitation source.The PXRD patterns were obtained on a Bruker D8-Advance X-ray diffractometer with CuKαradiation(λ=0.154 2 nm,U=40 kV,I=40 mA) in a 2θrange of 5°-50° at room temperature.The solid-state circular dichroism (CD) spectra were recorded on a JASCO J-810 spectropolarimeter with KCl pellets.UV-Vis spectra were acquired with a Shimadzu 2501 spectrometer(Japan).CV and DPV measurements were carried out on a CHI660 electrochemical workstation (Shanghai Chen Hua Instruments, China) at room temperature.A traditional three-electrode system consisting of a glassy carbon electrode (GCE) as the working electrode, a saturated calomel electrode (Ag/AgCl) as the reference electrode, and a platinum wire as the counter electrode was used.
1.2 Synthesis
1.2.1 Synthesis of compound 1-D
CoCl2·6H2O (0.047 6 g, 0.2 mmol),D-Hhpg(0.016 7 g,0.1 mmol),and distilled water (10 mL)were mixed and kept stirring until completely dissolved, and then the methanol solution (6 mL) with 4, 4′-bipy(0.015 6 g, 0.1 mmol) was added to the above solution and kept stirring for 20 min.The resulting solution was filtered and the filtrate was slowly evaporated at room temperature for 5-7 d or so to deposit red flaky crystals of compound 1-D with 55% yield based on Co.Anal.Calcd.for C18H20N3ClCoO5(%): C, 47.75; H, 4.45; N,9.28.Found(%): C, 47.18; H, 4.44; N, 9.38.IR (KBr,cm-1): 3 364 (s), 3 318(w), 1 608 (s), 1 589 (s), 1 498(m), 1 413 (s), 1 212 (m), 1 133 (s), 1 055 (m), 1 010(m),808(s),763(w),731(m),620(s),575(w).
1.2.2 Synthesis of compound 1-L
The synthesis method of 1-L was the same as 1-D,and the difference was to replaceD-Hhpg withL-Hhpg.Compound 1-L is red flaky crystals that were produced in a yield of 61% based on Co.Anal.Calcd.for C18H20N3ClCoO5(%): C,47.75; H,4.45; N,9.28.Found:C, 47.09; H, 4.39; N, 9.80.IR (KBr, cm-1): 3 363 (s),3 305(w), 1 615 (s), 1 562 (s), 1 504 (m), 1 411 (s),1 219(m),1 099 (s),1 045 (m),1 017(m),818(s),765(w),735(m),620(s),575(w).The XPS spectra of Co2pare shown in Fig.S1 (Supporting information).For compound 1-L, there were two main peaks corresponding to Co2p3/2and Co2p1/2at 780.82 and 796.36 eV,respectively.The peaks at 785.60 eV and 802.34 eV are the satellite peaks of Co2p3/2and Co2p1/2, respectively.According to bond valence calculation (BVS) and the law of conservation of charge, the central metal ion in compound 1-L is Co(Ⅱ).
1.2.3 Synthesis of compound 2-D
CoCl2·6H2O (0.047 6 g , 0.2 mmol),D-Hhpg(0.016 7 g, 0.1 mmol), and distilled water (8 mL) were mixed and kept stirring until completely dissolved, and then the ethanol solution (8 mL) with 5,5′-BM-2,2′-bipy(0.018 4 g,0.1 mmol)was added to the above solution and kept stirring for 20 min.The resulting solution was filtered and the filtrate was slowly evaporated at room temperature for two weeks or so to deposit red block crystals of compound 2-D with 65% yield based on Co.Anal.Calcd.for C28H39ClCoN4O11.5(%): C, 47.37;H, 5.54; N, 7.89.Found(%): C, 47.46; H, 5.78; N,7.86.IR (KBr, cm-1): 3 437 (s), 3 193(m), 3 084(w),1 662 (s), 1 513 (m), 1 475 (m), 1 347 (m), 1 244 (s),1 173(w),1 046(w),834(m),724(w),625(w),574(m).The XPS spectra of Co2pare shown in Fig.S1.For 2-D,there were two main peaks corresponding to Co2p3/2and Co2p1/2at 781.01 and 796.49 eV, respectively.The peaks at 784.80 eV and 800.44 eV are the satellite peaks of Co2p3/2and Co2p1/2, respectively.According to BVS and the law of conservation of charge, the central metal ion in compound 2-D is Co(Ⅲ).
1.2.4 Synthesis of compound 2-L
The synthesis method of 2-L was the same as 2-D,and the difference was to replaceD-Hhpg withL-Hhpg.Compound 2-L is red block crystals that were produced in a yield of 61% based on Co.Anal.Calcd.for C28H39ClCoN4O11.5(% ): C, 47.37; H, 5, 54; N, 7.89.Found(%): C, 47.25; H, 5.82; N, 7.95.IR (KBr, cm-1):3 430 (s), 1 661 (s), 1 519 (m), 1 448 (w), 1 450(w),1 357(m), 1 308 (w), 1 260 (s), 1 170(m), 1019 w), 827(w),584(m).
1.2.5 Preparation of working electrodes modified with compounds 1 and 2
Before each experiment, the GCE (diameter: 3 mm) was polished on a polishing pad with 0.3 and 0.05µm alumina powders.The cleaned GCE was dried with a nitrogen stream for the next modification.20.0 mg (or 12 mg) crystals were first mixed and ground with carbon black under mass ratios of 1∶3,and then dispersed into 200µL of 0.5% Nafion solution and ultrasonicated for 120 min to obtain a uniformly dispersed solution.Then, 10 µL catalyst suspension was dropped on the surface of GCEs and dried for testing.The Nafion solution acts as an adhesive and the protecting agent to ensure that the hybrid catalysts can be tightly modified on the surface of the GCE.
1.3 X-ray crystallography
Suitable single crystals for compounds 1 and 2 were selected for single-crystal X-ray diffraction analyses.The data for 1 and 2 were collected on a Bruker SMART-CCD diffractometer with MoKαradiation source(λ=0.071 073 nm)byφ-ωscan mode.The structures were solved through direct methods using SHELXS-97 and all non-hydrogen atoms were refined anisotropically by full-matrix least-squares onF2using SHELXL-97[33-34].Solvent removal can be carried out by using the SQUEEZE program.Further crystallographic data and experimental details for structural analyses of compounds 1 and 2 are summarized in Table 1.The selected bond lengths and angles of 1 and 2 are given in Table S1.Hydrogen bond lengths and bond angles are collated in Table S2.
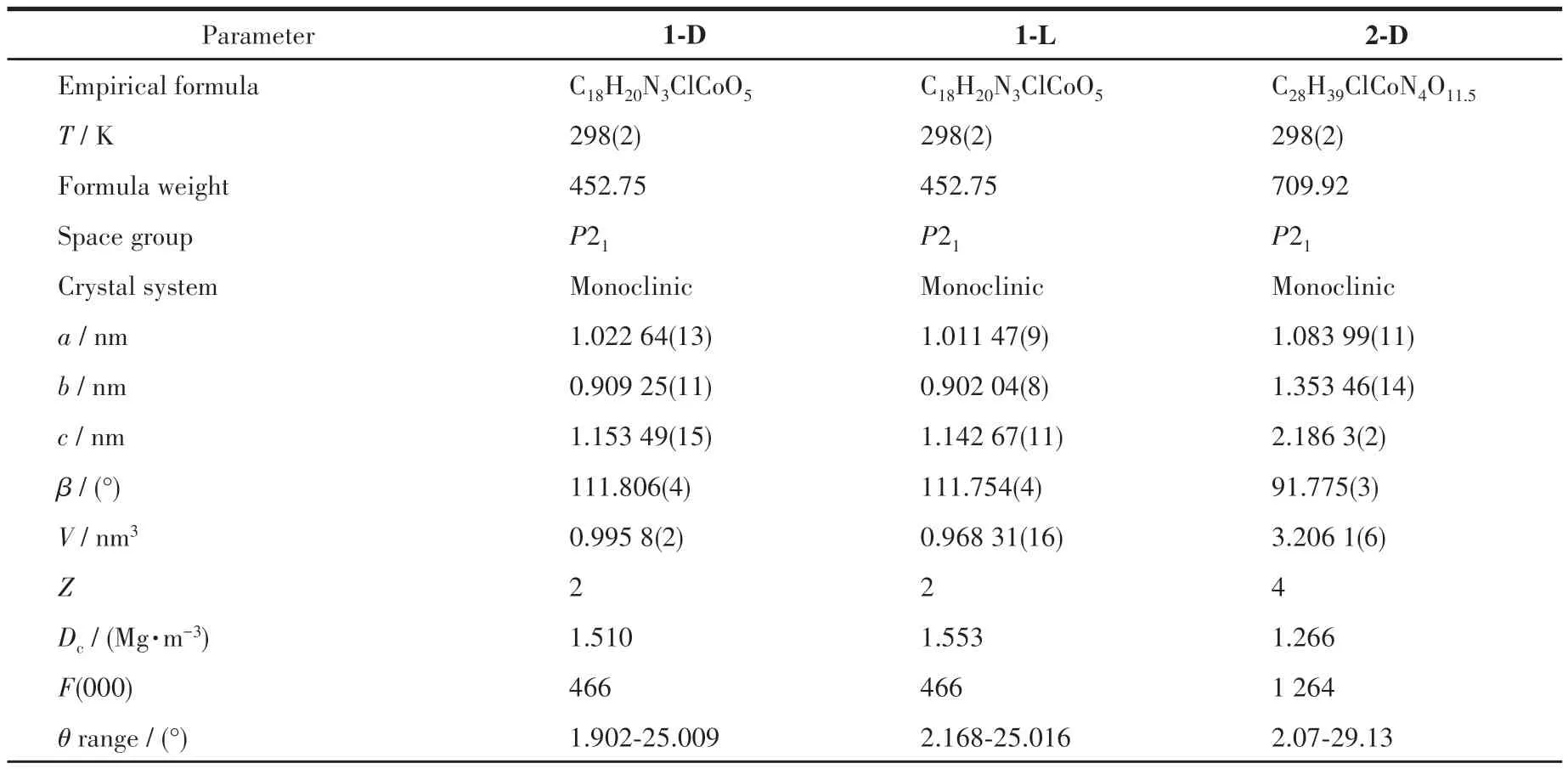
Table 1 Crystal data and structure refinements of the compounds
CCDC: 2124681, 1-D; 2124682, 1-L; 2124683,2-D.
2 Results and discussion
2.1 Structure description
2.1.1 Structural description of compounds 1-D and 1-L
Single crystal X-ray diffraction analysis revealed that compound 1 - D crystallizes in the monoclinic space group ofP21and possesses a 2D rectangular grid network.As shown in Fig.1A, the asymmetric unit is composed of one Co(Ⅱ)cation,oneD-hpg-anion,one 4,4′-bipy ligand, one Cl-counter anion, one coordination water molecule, and one lattice water molecule.The Co(Ⅱ)center is coordinated by three oxygen atoms and three nitrogen atoms to form a distorted octahedron geometry (CoO3N3).O1, O2#1, N1 are from twoD-hpgligands,N2,N3 are from two 4,4′-bipy auxiliary ligands,O4 is from a coordinated water molecule, where the octahedral equatorial positions are O1, O2, O4 and N1,N2 and N3 are in the axial positions of the octahedron.The Co—O bond lengths are 0.205 5(14)-0.213 0(11)nm and the Co—N distances are 0.218 1(15)-0.222 0(11)nm, respectively.All of which fall within the normal ranges[35].
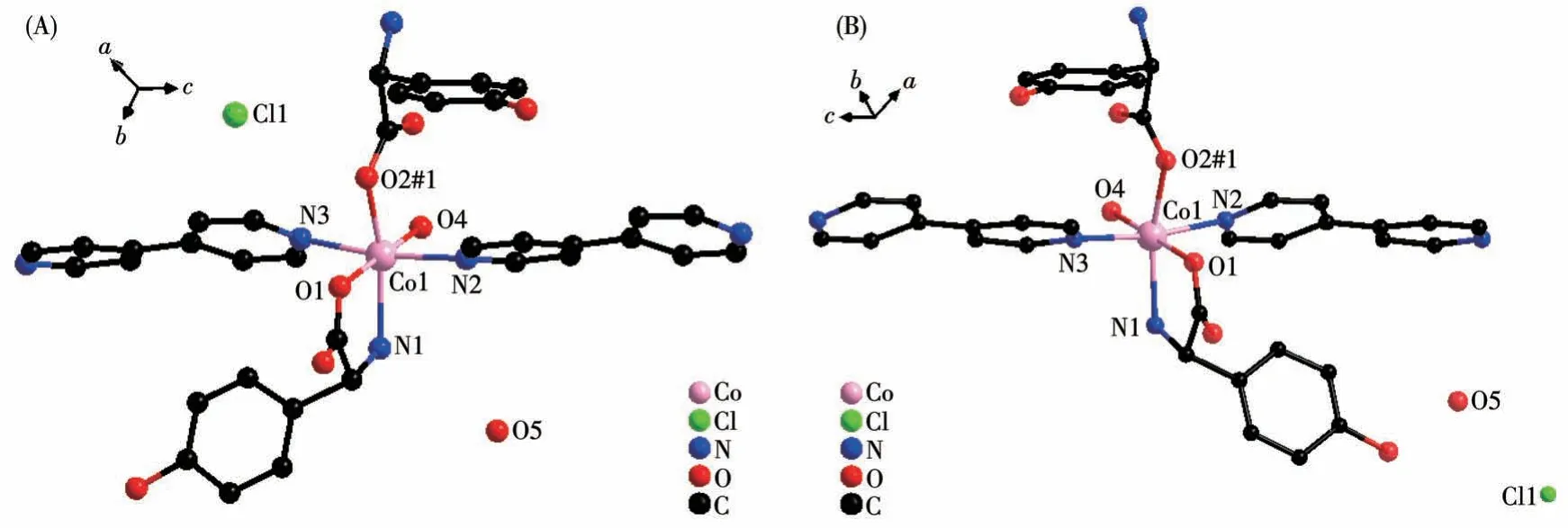
Fig.1 Coordination environment of Co(Ⅱ)in compounds 1-D(A)and 1-L(B)
TheD-hpg-adopts as aμ2-η1∶η1∶η1coordination mode (two carboxyl oxygen atoms and one amino nitrogen atom) to connect the two metal cobalt, forming a 1D right-handed helical chain structure along thea-axis (Fig.2A).In addition, these chains are further linked via 4,4′-bipy molecules to build a 2D (4,4)rectangular grid network parallel to the bc plane, and the size of the rectangular grid is 0.519 nm×1.153 nm,respectively, as shown in Fig.3A.There are extensive hydrogen bonds among carboxyl oxygen atoms(O1,O2#7), amino nitrogen atom (N1), coordinated water molecule (O4) and free Cl-anion, which further connects the 2D networks to form a 3D supramolecular structure, as depicted in Fig.4A.The typical hydrogen bonds are N1…O3#5 0.319 0(18) nm, N1…Cl1#6 0.340 0(13) nm, O3…O5#4 0.272(2) nm, O4…O1#1 0.275 3(16)nm,O4…O2#7 0.335 2(16)nm,O5…Cl1#6 0.320 3(18) nm (Symmetry codes: #1: -x+1,y-1/2,-z+1;#3:-x+1,y+1/2,-z+1;#4:x,y,z-1;#5:-x,y-1/2,-z;#6:x-1,y,z;#7:x,y-1,z).
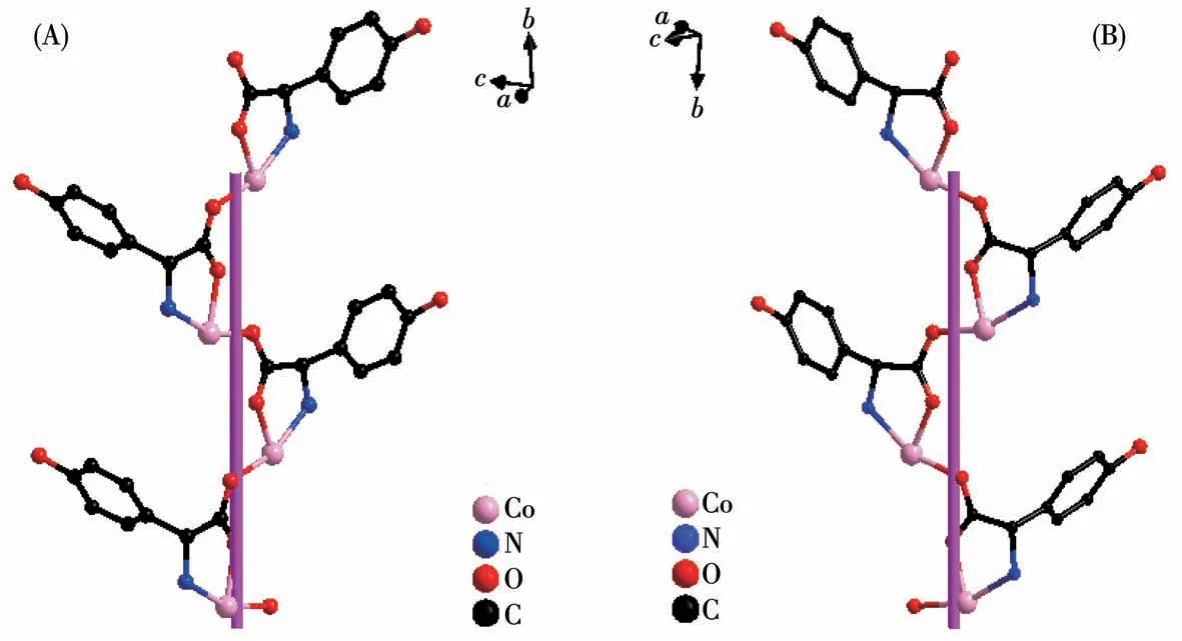
Fig.2 (A)One-dimensional right-handed helical chains of compound 1-D;(B)1D left-handed helical chains of compound 1-L
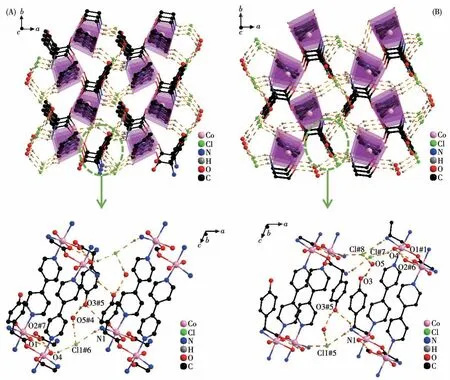
Fig.4 Three-dimensional supramolecular framework of compounds 1-D(A)and 1-L(B)
By comparing the crystallography data of 1-D and 1-L shown in Table 1 as well as Fig.1A and 1B, we conclude that compounds 1-D and 1-L are enantiomers.In compound 1-L, theL-hpg-anion adopts theμ2-η1∶η1∶η1coordination mode to connect adjacent Co ions.Compared with compound 1-D,1-L forms 1D lefthanded helical chains (Fig.2B).These chains are further linked via 4,4′-bipy molecules to build a 2D (4,4)rectangular grid network parallel to thebcplane(Fig.3B), and the size of the rectangular grid is 0.519 nm×1.142 nm, respectively, and 1-L eventually forms a 3D supramolecular structure through hydrogen bond interactions, as depicted in Fig.4B.The typical hydrogen bonds are N1…Cl1#5 0.335 0(13) nm, N1…O3#5 0.313 1(17) nm, O3…O5 0.267(2) nm, O4…O1#1 0.270 8(16)nm,O4…O2#6 0.332 2(15)nm,O4…Cl1#7 0.309 5(12)nm (Symmetry codes:#1:-x+1,y+1/2,-z+2;#5:-x,y+1/2,-z+1;#6:x,y+1,z;#7:x,y+1,z+1).
2.1.2 Crystal structure of compound 2-D
Single crystal X-ray diffraction analysis revealed that compound 2-D crystallizes in a monoclinic space group ofP21and possesses a 0D small molecule structure.As shown in Fig.5, the asymmetric unit is composed of one Co(Ⅲ)cation,twoD-hpg-anions,one 5,5′-BM-2,2′-bipy ligand, one Cl-counter anion, and 5.5 lattice water molecules.Each Co(Ⅲ)is six-coordinated with the geometry of distorted octahedral geometry,which is completed by O1, N1, N2, O4 from twoDhpg-ligands and N3, N4 from one 5,5′-BM-2,2′-bipy ligand.The O1,O4,N2,and N3 atoms form the equatorial plane, while the N1 and N2 atoms are located in the axial positions.The Co—O bond lengths are 0.184 2(7) and 0.187 8(6) nm, and the Co—N bond lengths range from 0.188 2(8) to 0.193 7(9) nm, which are in accordance with the previously reported compounds[35].
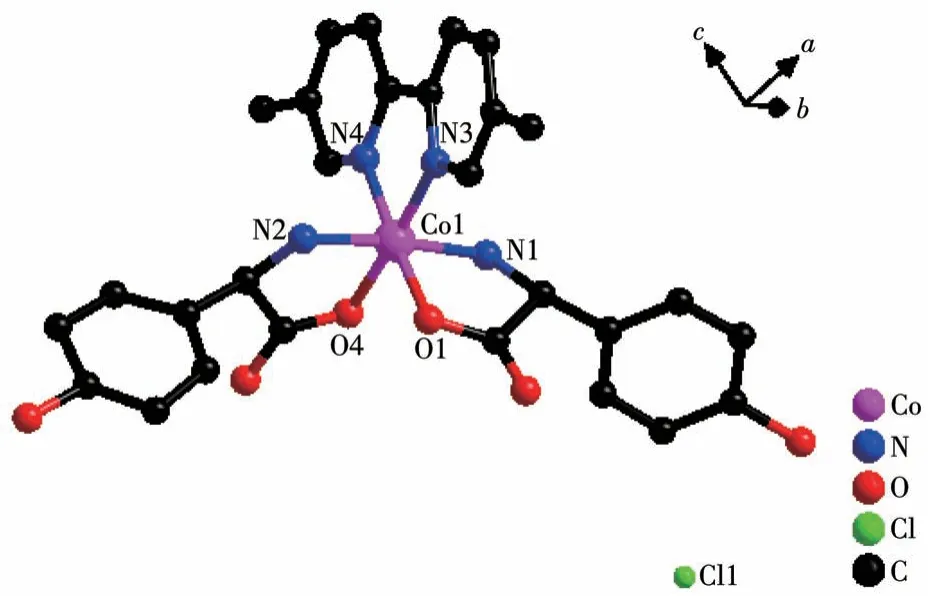
Fig.5 View of coordination environment of Co(Ⅱ)in compound 2-D
Both the twoD-hpg-anions and 5,5′-BM-2,2′-bipy ligands act as bidentate ligands with chelating mode (one carboxyl oxygen atom and one amino nitrogen atom, two pyridine nitrogen atoms) to coordinate with the metal cobalt ions, so each asymmetric unit contains three penta-atomic chelate rings.However,adjacent small molecules are connected by hydrogen bonds between the carboxyl oxygen atoms and amino nitrogen atoms of theD-hpg-ligand to form a dimer unit.These dimer units form a 1D supramolecular double chain along theb-axis through the hydrogen bonds between the hydroxyl oxygen atom and the amino nitrogen atom of theD-hpg-ligand (the hydrogen bonds:N2…O8#3 0.297 8(11) nm, N6…O(2)#1 0.284 2(11)nm,N1…O9#1 0.311 0(12)nm,N5…O3#3 0.295 4(13)nm), as shown in Fig.6.In addition, the 1D double chains are arranged in the form of ABAB stacking in the a-axis direction, as shown in Fig.7.The typical hydrogen bonds are N1…O9#1 0.311 0(12) nm, N1…Cl2#2 0.330 2(8)nm,N2…O8#3 0.297 8(11)nm,N5…Cl1# 0.319 1(9) nm, N5…O3#3 0.295 4(13) nm, N6…O2#1 0.284 2(11) nm, O9…N1#3 0.311 0(12) nm(Symmetry codes: #1: -x,y+1/2, -z+1; #2:x-1,y,z;#3:-x,y-1/2,-z+1).
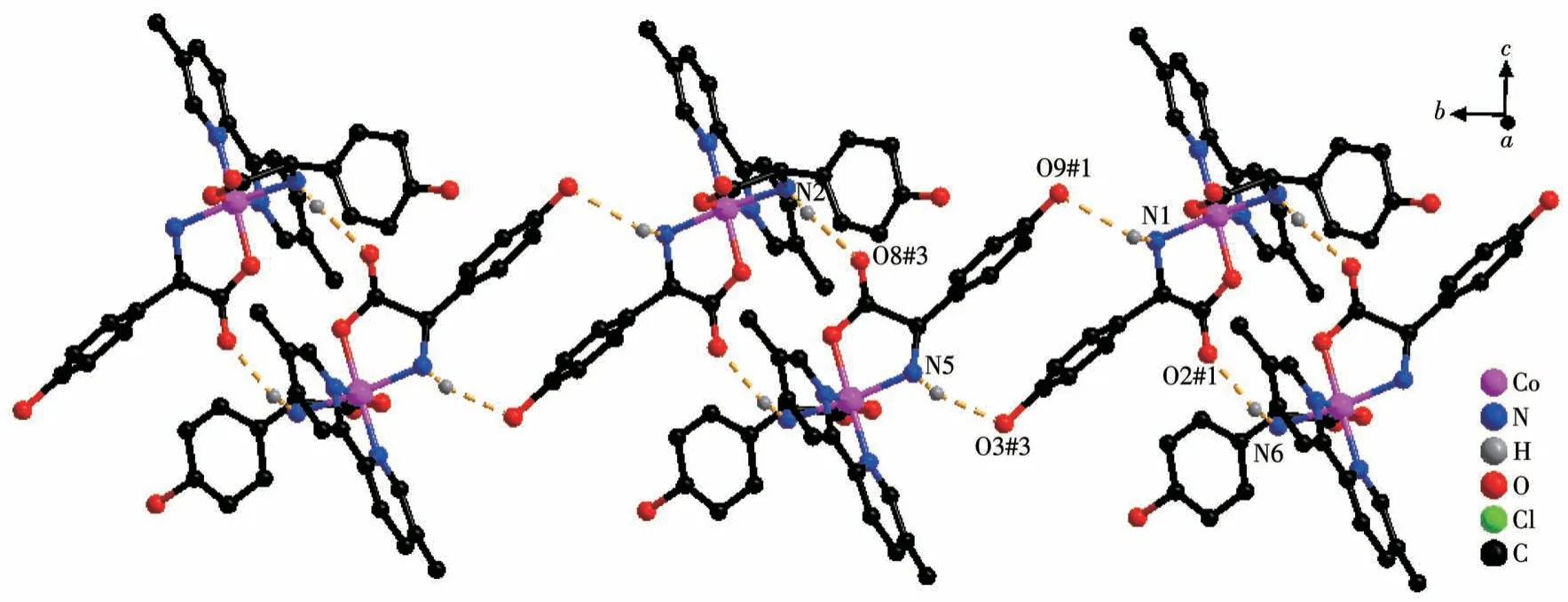
Fig.6 One-dimensional supramolecular double chain of compound 2-D
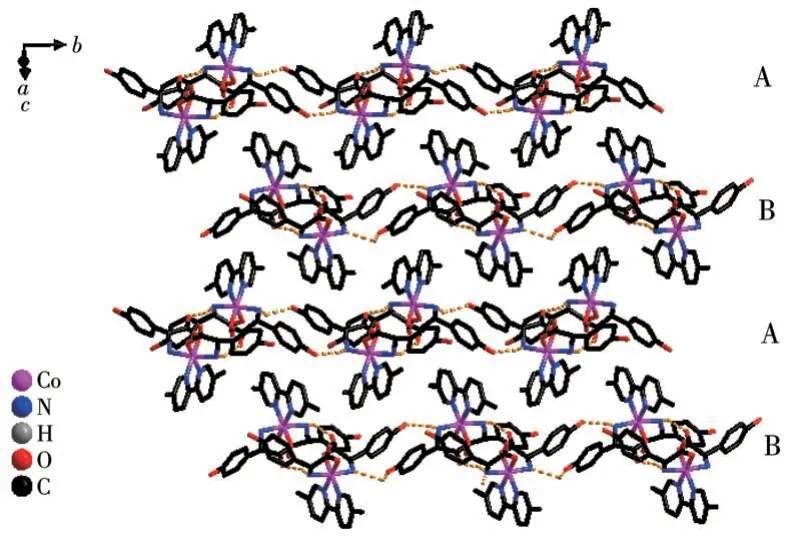
Fig.7 Chains arranged in ABAB fashion along the a-axis in compound 2-D
2.1.3 Comparison of the structures
From the structure descriptions above, we found the different auxiliary ligands(4,4′-bipy,5,5′-BM-2,2′-bipy) affected the coordination mode of the organic ligands, further, the structure of compounds 1 and 2 were transformed (Scheme 1).When the 4,4′-bipy molecule is selected as the auxiliary ligand in 1, theD/Lhpg-exhibits theμ2-η1∶η1∶η1coordination mode and bridges the more neighboring Co(Ⅱ)cations and extends infinitely along theb-axis direction to form a 1D helical chain.These 1D chains are further linked via 4,4′-bipy molecules to build a 2D rectangular grid network.When 5,5′-BM-2,2′-bipy is used instead of 4,4′-bipy in 2-D,theD-Hhpg andL-Hhpg ligands exhibit chelating bidentate coordination patterns with Co (Ⅲ)ions.Moreover,the 5,5′-BM-2,2′-bipy molecule also participates in the coordination of compound 2-D through two pyridine nitrogen atoms in a chelated bidentate mode,which does not act as a bridge, and the compound finally presents a 0D molecular structure.In short,different N-donor ligands induce a shift in the coordination mode of Hhpg, which in turn reduces the dimensionality of the compounds from a 2D rectangular grid network to a 0D molecule structure.Therefore, the formation of compounds with different structures is wellcontrolled by modulating N-donor ligands, which may provide us with a simple and efficient synthetic route for the tunable construction of compounds.
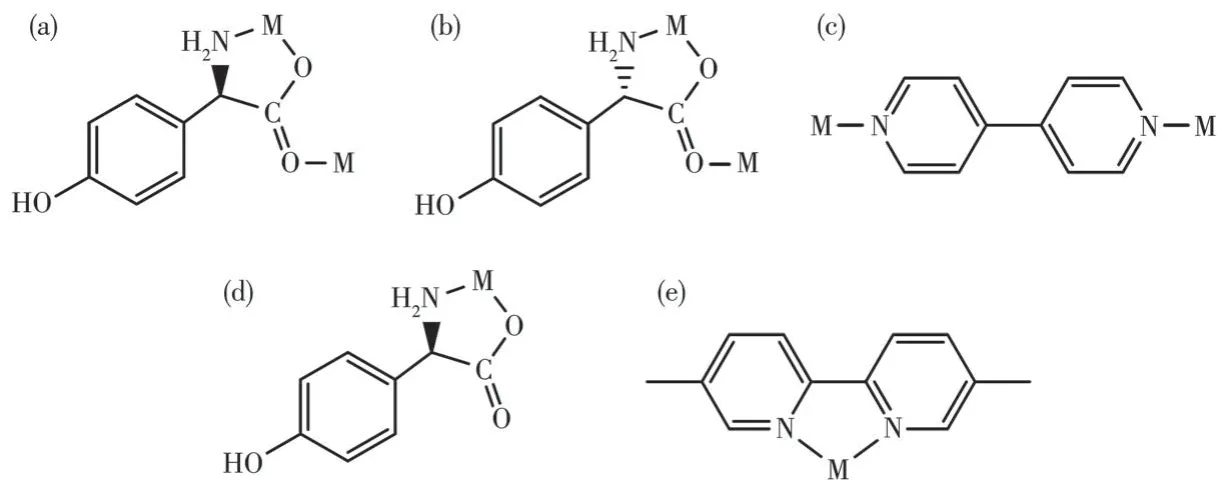
Scheme 1 Observed coordination mode of the ligands:(a)D-Hhpg ligand for compound 1-D;(b)L-Hhpg ligand for compound 1-L;(c)4,4′-bipy ligand for compounds 1-D and 1-L;(d)D-Hhpg ligand for compound 2-D;(e)5,5′-BM-2,2′-bipy ligand for compound 2-D
2.2 Solid-state CD spectra
To further demonstrate the homochirality of compounds 1 and 2, solid-state CD analysis has been conducted.As shown in Fig.8, the CD curve ofD-Hhpg displayed a positive Cotton effect with peaks at 249 and 271 nm, while theL-Hhpg displayed a negative Cotton effect at 248 and 272 nm.Compound 1-D displayed positive peak values at 253 and 262 nm.Compound 1-L displayed negative peak values at 252 and 263 nm.Compound 2-D displayed positive peak values at 260 and 290 nm.Compound 2-L displayed negative peak values at 261 and 288 nm.Compounds 1-D/1-L(2-D/2-L)had the opposite CD signal in the same peak position.The result confirms that the 1-D/1-L and 2-D/2-L are homochiral compounds.

Fig.8 Solid-state CD spectra of D-Hhpg,L-Hhpg,and compounds 1 and 2
2.3 Thermal analyses
The thermal behaviors of compounds 1 and 2 were investigated under a dry nitrogen atmosphere from 20 to 800 ℃and the TG curves are presented in Fig.S2.1-D and 1-L had similar thermochemical behaviors,and two weight-loss steps were observed in Fig.S2a and S2b, due to the continuous loss of crystal water, 4,4′-bipy as well as the collapse of crystal structure,respectively.The results show that the main structure of the compounds 1-D/1-L was stable up to 139 ℃/152 ℃.For 2-D(2-L),the first weight loss of 10.16%(11.17%)happened at 31.0-151.0 ℃(31.0-154.0 ℃), which corresponds to the removal of four lattice water molecules per formula unit (Calcd.10.55%).From 151.0 to 295.0 ℃(from 154.0 to 284.0 ℃), the second weight loss of 19.80% (18.39%) corresponds to one and a half lattice water molecules and part 5,5′-BM-2,2′-bipy molecule.Beyond 295.0 ℃(284.0 ℃), a rapid weight loss was observed, indicating the rapid decomposition of compound 2-D (2-L).Compounds 1 and 2 continued to lose weight up to 800 ℃, indicating the continuous expulsion of organic moieties even at the upper limit of the measurement range.
2.4 PXRD analyses and IR spectra
To check the phase purity of the crystals, the PXRD patterns for compounds 1 and 2 were presented in Fig.S3.The main peaks of simulated patterns of 1-D, 1-L, 2-D, and 2-L match well with their experimental patterns, demonstrating the crystallization degree and the purity of the crystalline phase both are good.
From the IR spectra of compounds 1 and 2 illustrated in Fig.S4, we found that the characteristic peaks of enantiomer compounds were almost completely identical.Theνas(COO-) andνs(COO-) vibrations ofD-hpg-/L-hpg-could be observed at 1 601-1 655 cm-1and 1 410-1 513 cm-1, respectively.The peak at 1 224-1 251 cm-1is due toν(C—O) vibrations of the phenolic hydroxyl group, and the peak at 1 309-1 378 cm-1is due toνas(C—N) vibrations of amino groups which connect to the carbon atoms in amino-acids.Moreover, theν(C—C)andν(C=N)bending vibration of the pyridine ring at 1 517 and 1 311 cm-1, indicates that the 4,4′-bipy molecules have coordinated with metal in compounds 1-D and 1-L[36].Theν(C—C) andν(C=N)bending vibration of the pyridine ring at 1 481-1 485 cm-1and 1 311-1 323 cm-1indicates that the 5,5′-BM-2,2′-bipy molecules have coordinated with metal in compounds 2-D and 2-L[37].
2.5 Electrochemical properties of compounds 1 and 2
Electrochemical redox properties of compounds 1 and 2 were studied by using CV methods in dilute H2SO4solution via a three-electrode system.To promote the electroconductivity of the modified electrode,carbon black was employed.The selected mass ratio was 3∶1 for carbon black and solid crystal, in which the largest electrochemical area and smallest impedance were found (Fig.S5).As shown in Fig.9A and 9B,two pairs of reversible redox peaks (labeled as Ⅱ-Ⅱ′,Ⅲ-Ⅲ′) appeared in the range of the tested potential.The peak potentials of Ⅱ-Ⅱ′and Ⅲ-Ⅲ′were 0.551 V(Ⅱ)/0.363 V(Ⅱ′) and -0.468 V(Ⅲ)/-0.403 V(Ⅲ′) for GCE-1-D, and 0.476 V(Ⅱ)/0.337 V(Ⅱ′), -0.512 V(Ⅲ)/-0.382 V(Ⅲ′) for GCE-1-L, respectively.A pair of reversible redox peaks (labeled Ⅱ-Ⅱ′) can be observed in Fig.9C and 9D.The peak potentials of Ⅱ-Ⅱ′are 0.449 V(Ⅱ)/0.335 V(Ⅱ′) for GCE-2-D, and 0.433 V(Ⅱ)/0.307 V(Ⅱ′) for GCE-2-L, respectively.The redox peak (Ⅱ-Ⅱ′) should be attributed to the redox process of Co (Ⅱ)/Co(Ⅲ), and the redox peak (Ⅲ-Ⅲ′)should be attributed to the redox process of Co (Ⅱ)/Co(0).The observed irreversibility of these reductions in GCE-1-L is proposed to be due to the decomposition of Co(0)to elemental Co,whose deposition on the working electrode may lead to the observed non-Faradaic current[38].Moreover, the redox peak currents of compounds 1 and 2 increased as the scan rate increased,and it was found that there was a positive correlation between peak currents and sweep speeds, indicating the surface-controlled feature of the redox process.It is noteworthy that the peak position remained unchanged with the increase in scan speed, indicating the excellent and stable electrochemical redox properties of compounds 1 and 2.

Fig.9 CV curves of compounds 1 and 2 with different scan rates in 0.5 mol·L-1 H2SO4 solution
2.6 Electrochemical chiral recognition of His enantiomers via CV technique
The interaction between His enantiomers and the chiral interface was measured in 1.0 mmol·L-1L-His orD-His (40 mL, 0.5 mol·L-1Na2SO4) solution via CV technique at the potential range from -1 to 1.2 V.As shown in Fig.10, the bare GCE behaved low and overlapped currents forL-His andD-His, which means no ability of chiral recognition by bare GCE.Significantly increased peak currents of His enantiomers were obtained on GCE-1/2 due to the excellent electrocatalytic properties of compounds 1 and 2.However, the simultaneous shift of peak potential on the other working electrodes resulted in little potential difference for the two enantiomers, indicating hardly to meet the requirement for dual - enantiorecognition.Compared with other working electrodes, the larger peak electric potential difference toL-His andD-His was acquired on GCE-1-D,revealing that 1-D can provide a delicate chiral environment in the enantioselective interaction.These suggest that the chiral compound 1-D has outstanding conductivity and different electrocatalytic functions on the oxidation ofD-/L-His.Based on the facts,the strategy for enantiodiscrimination ofD-/L-His enantiomers with the chiral compound 1-D material can be achieved.
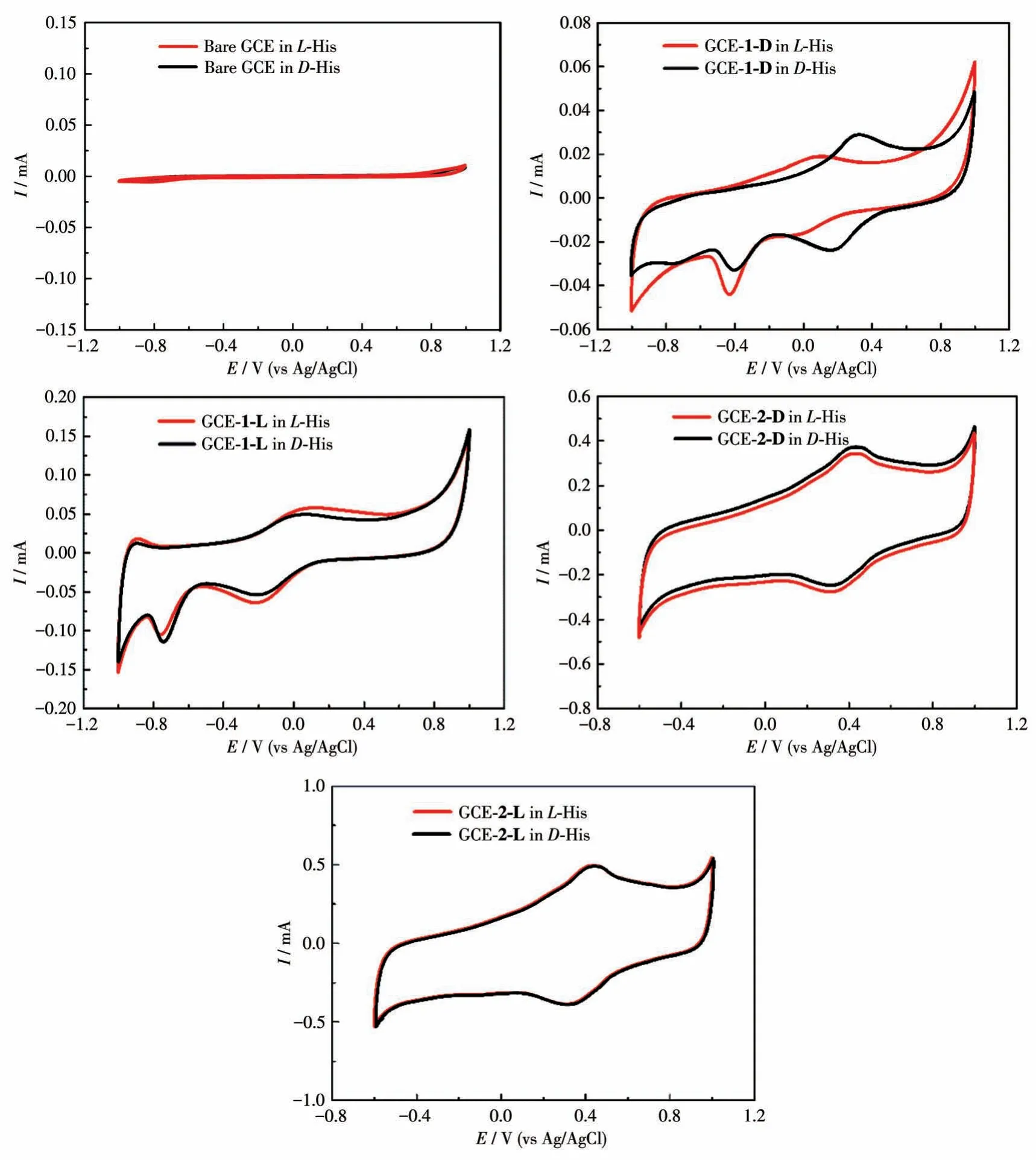
Fig.10 CV curves of different modified electrodes for the detection of 1.0 mmol·L-1 D-/L-His in 0.5 mol·L-1 Na2SO4
2.7 Optimization of the experimental condition
To explore the optimal condition for the enantioselective efficiency of GCE-1-D, we have studied the influence of pH on the peak current ratio betweenL-His andD-His (Fig.S6).As shown in Fig.11A, we can easily find that the chiral recognition efficiency(difference of peak potential) gradually increased with the pH ranging from 4.0 to 5.0, and the maximum efficiency at pH=5.0.These anodic peak potentials ofLHis andD-His at pH=6.0 or pH=7.0 were not suitable for assaying the enantiomers owing to their weak peak currents.Therefore, pH=5 was chosen as the optimum pH for further chiral recognition measurements.
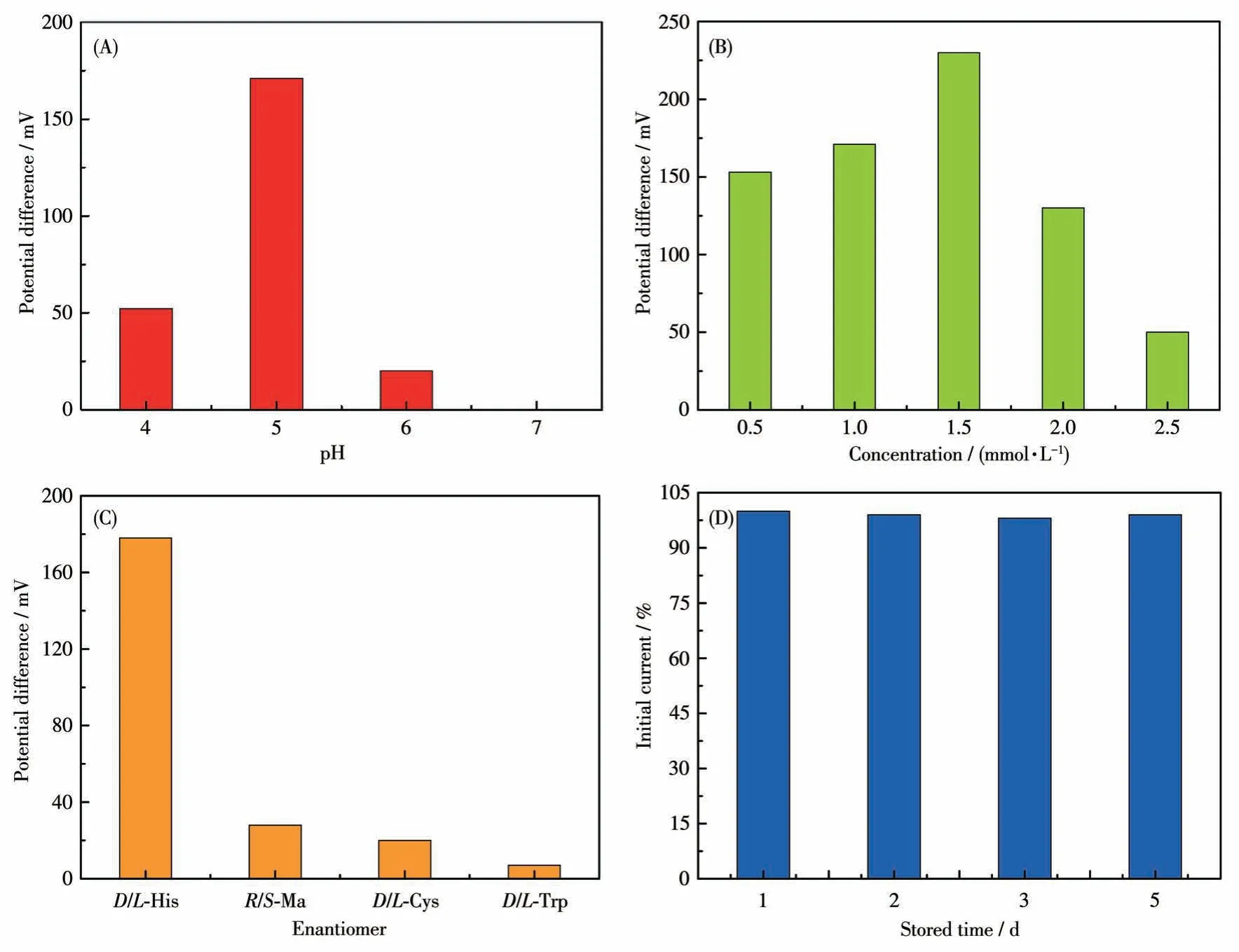
Fig.11 (A)Recognition efficiency of GCE-1-D for His isomers in different pH values via CV technique;(B)Recognition efficiency of GCE-1-D in different concentrations of His isomers;(C)Enantioselective interaction with the His,Cys,Ma,and His on GCE-1-D via CV technique;(D)Stability of the GCE-1-D electrode for L-His in different stored times
Under optimal pH of 5, a series of His enantiomers with different concentrations were investigated using the GCE-1-D(Fig.S7a-S7e).Fig.11B showed that the chiral recognition efficiency (ΔEp=Ep,D-Ep,L) of GCE gradually increased with the increasing concentration ofL-His andD-His.Increasing concentration of His isomers from 0.5 to 1.5 mmol·L-1,the chiral recognition efficiency gradually increased and reached the maximum value at 1.5 mmol·L-1, which may be attributed to a state of exactly saturated adsorption, and more than 1.5 mmol·L-1His isomers might cause the decreasing conductivity of the electrolyte solution.Therefore, 1.5 mmol·L-1was the optimal concentration of His isomers in this chiral recognition experiment.In addition, the peak potential of GCE-1-D onD-His was greater than that onL-His in this concentration range(Fig.S7f), indicating that the chiral compound-modified electrodes can selectively interact withL-His andD-His.
2.8 Enantioselectivity and stability of the chiral interface
Amino acids enantiomers of cysteine (Cys),tryptophan (Trp), and mandelic acid (Ma) were respectively used to investigate the chiral specificity of the chiral interface (Fig.S8).The recognition efficiency of His was greatly higher than the other three amino acids enantiomers, hinting that this chiral interface possessed high enantioselectivity for His enantiomers (Fig.11C).Moreover, the peak currents decreased less than 2% compared to the initial response ofL-His after the modified electrodes were stored for 5 d at 4 ℃(Fig.11D).These results demonstrate that this method possesses acceptable enantioselectivity and stability.
2.9 Electrochemical chiral recognition of His enantiomers via DPV technique
DPV is a powerful method in electrochemical analysis because of its higher sensitivity.Therefore, to further confirm the ability of compound 1-D to recognize His enantiomers, DPV was used to verify its electrochemical recognition performance.Using the same electrode and equipment as the CV technology in the electrolyte solution (40 mL, 0.5 mol·L-1Na2SO4), the DPV curves for identifying His enantiomers under different pH values were recorded at a scan rate of 50 mV·s-1.The maximum chiral recognition ability was obtained at pH=5 as shown in Fig.12A.The recognition results from DPVs were consistent with the conclusions of CVs, which demonstrates that the electrode modified by compound 1-D has better electrochemical recognition performance.And under the optimal pH condition, we continue to explore the influence of different concentrations on the recognition efficiency (Fig.S9 and S10).The modified electrode displayed the best recognition effect on 1.0 mmol·L-1His enantiomers.The electrochemical test results show that the electrode modified by compound 1-D can be used for the recognition of His enantiomers as a promising chiral material to construct electrochemical sensing platforms.
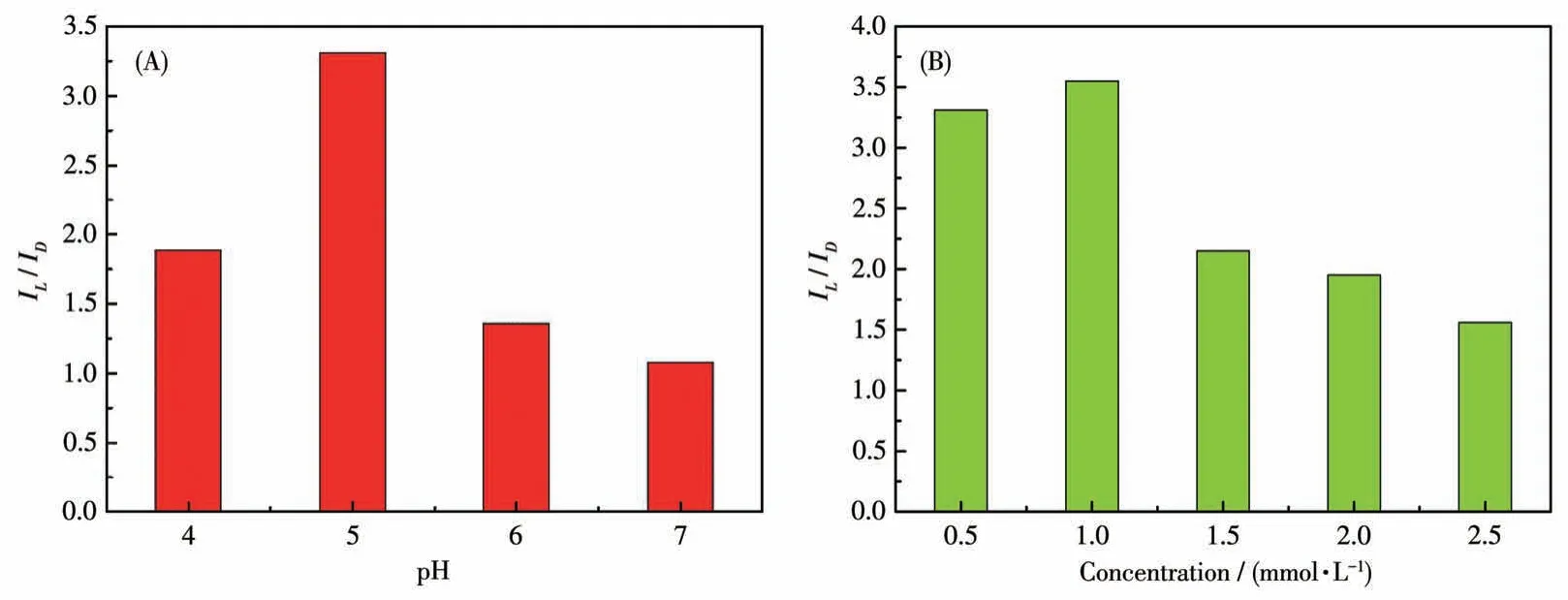
Fig.12 (A)Recognition efficiency(IL/ID)of GCE-1-D for His isomers in different pH values via DPV technique;(B)Recognition efficiency of GCE-1-D in different concentrations of His isomers via DPV technique
2.10 Identification of D-/L-His in racemic mixture
The performance of the chiral sensor to identify the ratio ofL-/D-His enantiomers in the racemic solution is significant for further practical applications.The proposed GCE-1-D electrochemical sensor was applied to determine the precise mass fraction ofD-His (wD-His)in a racemic mixture with 1 mol·L-1total concentration.As shown in Fig.13A, the oxidation peaks ofD-His at about 13.5 mV andL-His at about 44.5 mV(Fig.S9b) were combined into a single one, which gradually shifted in the positive direction asD-His concentration increased.More importantly, the oxidation currents againstwD-Hisin the mixture of His enantiomers exhibited a linear dependence aswD-Hisincreased with the correlation coefficient of 0.922 8 (Fig.13B).These results imply that the GCE-1-D chiral sensor can effectively quantify the ratio of His enantiomers for a racemic sample from a practical point of view.From the perspective of practical application, this has a certain guiding significance for the accurate determination of the drug isomers in a racemic solution.
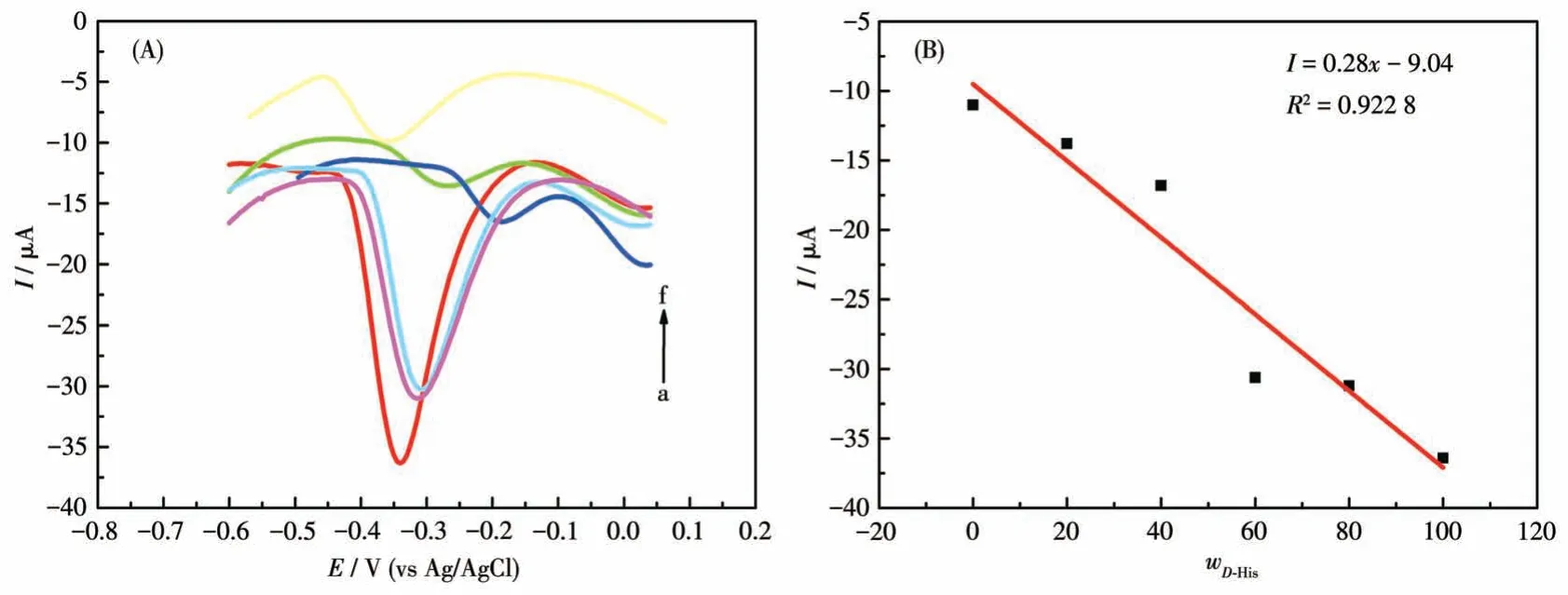
Fig.13 (A)DPV curves of GCE-1-D for obtaining wD-His in the mixture of His enantiomers;(B)Linear relationship between wD-His and the oxidation currents
3 Conclusions
By using chiral amino acid derivativesD-/L-Hhpg and metal Co ion in the presence of N-donor ancillary ligands, two pairs of enantiomeric coordination compounds have been successfully prepared.Compounds 1-D and 1-L crystallize in the chiral monoclinic space groupP21and show 2D (4,4) rectangular grid networks that consist of left- or right-handed helical chains.Compound 2-D belongs to theP21space group of the chiral monoclinic system and exhibits 0D molecule structures.Compounds 2-D and 2-L are enantiomers.Furthermore, compound 1-D can serve as an electrochemical sensor for efficiently detecting His enantiomers and quantitatively determining the enantiomeric excess in the His mixture on account of its electrochemically reversible redox behavior.This work provides an economically cheap and easy-to-handle route for selective identification of chiral His enantiomers.
Acknowledgments:This work was supported by the Natural Science Foundation of Hebei Province(Grant No.B2018205152),the Project of Introduction of Overseas Returnee in Hebei Province (Grant No.CL201715), and the Key Project of the Natural Science Foundation of Hebei Normal University (Grant No.L2021Z03).
Supporting information is available at http://www.wjhxxb.cn
