Cu基催化剂电催化还原CO2的性能调控及其产物的研究进展
2024-01-15李好常少华张萌萌
李好,常少华,张萌萌
(天津工业大学材料科学与工程学院,天津 300387)
近年来,蓬勃发展的工业正在加速全球化石能源的消耗,大量燃烧化石能源势必造成大气污染和温室效应加剧。当前正处于能源变革的交叉路口,在化石能源枯竭和环境突变的背景下,迫切需要加强对CO2转化技术的研究。将气态形式的CO2收集并作为一种生产原料是缓解温室效应的有效手段。相比氢能,液体的碳能源更适于人类利用[1]。因此,利用CO2生产碳氢燃料的技术引起了人们的广泛关注。自1985年霍利发现可在Cu电极上利用电还原的方式将CO2转化为碳氢化合物[2],越来越多的研究人员尝试利用各种化学手段转化CO2。基于此,本文将总结关于各类Cu基电催化剂的设计合成以及不同种类的碳还原产物。
1 电催化还原CO2概述
电催化还原CO2(eCO2R)反应涉及气、液、固三相,具体为气体原料、液态电解质和固态电极。从热力学角度而言,CO2在环境条件下是高度稳定的分子,C=O 双键的解离能可达750 kJ/mol,结构不易被破坏,在还原过程中很难被活化导致低的反应活性[3]。从动力学角度而言,eCO2R 为碳链产物的异相催化存在多种反应途径,主要存在以下几个过程:1)CO2在电催化剂表面的化学吸附,并通过与表面金属原子的相互作用形成一个具有部分电荷的*CO2δ-。值得注意的是,线性CO2分子也可能发生物理吸附。随着电解过程的进行,物理吸附的CO2可以通过表面缺陷、碱金属促进的表面或电子进一步激活为化学吸附形式。2)CO2吸附在催化剂表面后,将发生电子转移或质子耦合以切割C-O键,形成还原的中间体。3)C-C 键形成并产生多种产物,包括碳氢化合物和含氧物。4)重排碳产物,产物从活性位点解吸并扩散到电解质溶液中,即产物从催化剂表面脱附而生[4]。
CO2在吸附过程中主要有以下3种吸附模型[5]:1)氧配位模型:CO2分子作为电子供体,其每个氧原子都有一对单独的电子可以贡献给表面的路易斯酸中心,如图1(a)所示。2)碳配位模型:即带正电荷的碳原子作为电子受体接收来自表面路易斯碱的电子,如氧化物离子,形成如图1(b)所示的碳酸盐物种。3)混合配位模式:CO2分子中的碳原子和氧原子分别作为电子受体和供体,共同工作形成混合配位结构,如图1(c)所示。表面*CO2δ-的不同结合模式显著影响CO2的还原途径。通常,碳原子与路易斯碱基中心上的单齿酸结合有利于羧基(-COOH)的生成,其是CO 形成的关键中间体。两个氧原子在氧配位模式下的双齿结合,通过促进质子附着到CO2的碳原子上,形成甲酸盐阴离子。CO2还原过程中,步骤1 中假定的配位模式可能会显著影响后续的步骤2 和步骤3,导致形成甲酸盐、CO 和一系列C2产物的反应途径存在差异。eCO2R半反应的主要电极电势如表1所示[6-7]。
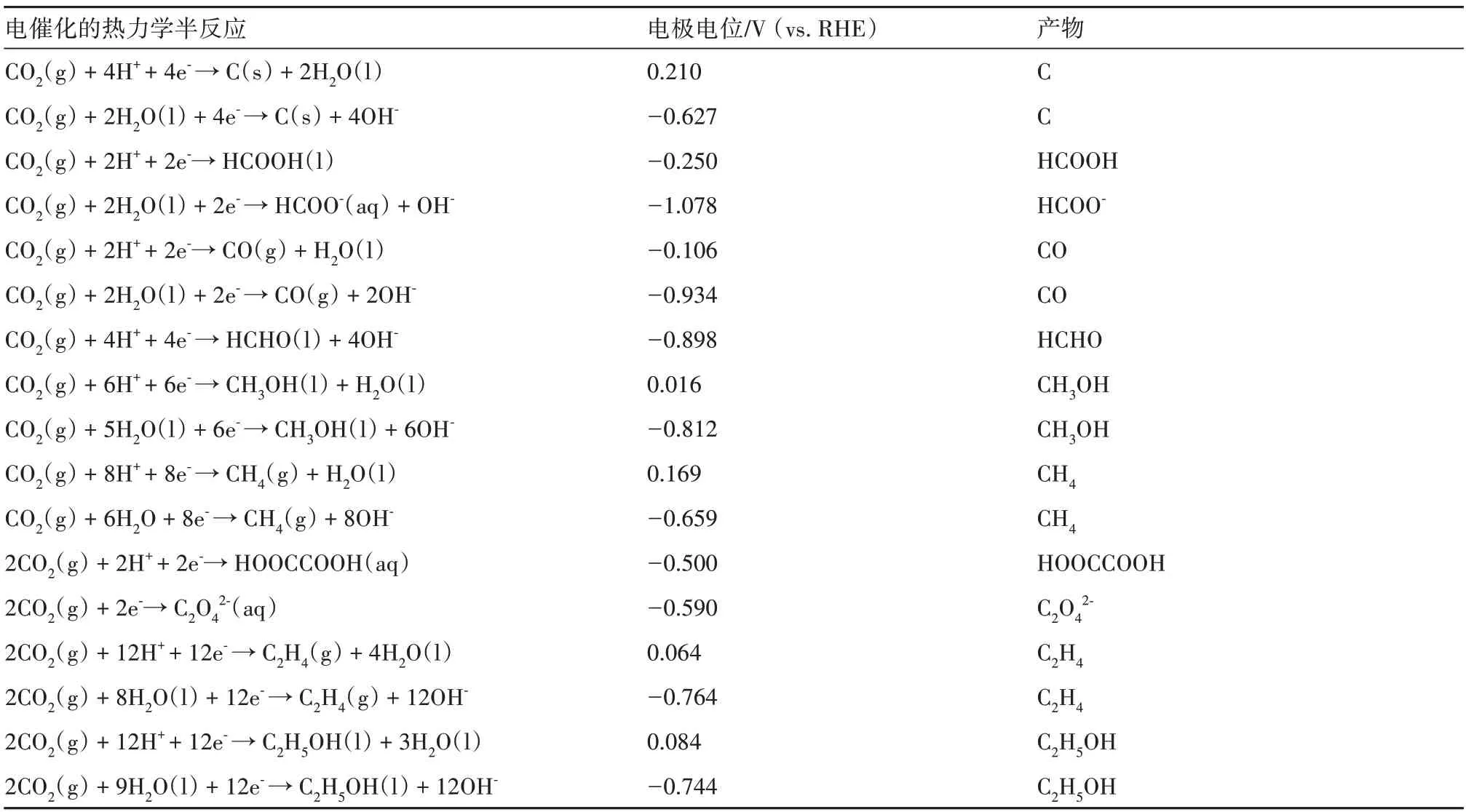
表1 标准条件下eCO2R半反应的电极电位及产物(1.01×105 Pa,25 ℃,pH值为7)Table 1 Electrode potentials and products of eCO2R half-reaction under standard conditions(1.01×105 Pa,25 °C,pH 7)[6-7]
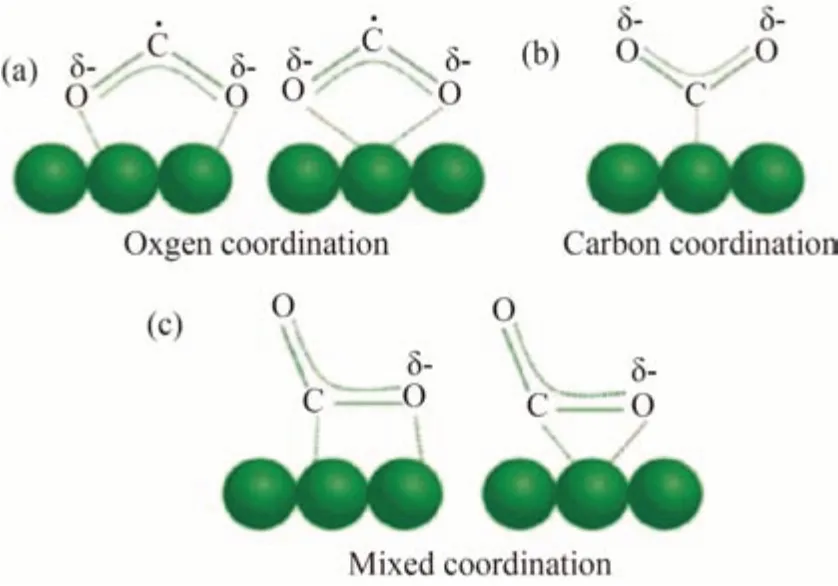
图1 CO2的吸附模型(a)氧配位模型;(b)碳配位模型;(c)混合配位模型Fig.1 CO2 adsorption models[5](a)Oxygen coordination model;(b)Carbon coordination model;(c)Mixed coordination model
综上可知,将CO2电催化还原为更复杂的C2产物之前,CO2可能先被还原为CO[8]。经过质子耦合电子的转移过程后,可能会形成羧基,随后羧基经过一系列的反应并改变CO 还原速率。CO2与活性位点吸附后生成CO 的路径如图2 所示[5]。通常,在使用特殊的催化剂和电解质时,eCO2R 反应可以通过质子化过程和水合反应两个过程进行[9]。这两个过程存在不同的反应机制和标准氧化还原电位。质子化过程通常需要比水合反应过程更低的电位,表明质子相关的电子转移过程在热力学上更有利于生成C2产物。然而,由于较高的能量势垒,C2产物的生成比C1产物更困难,通常需要更高的过电位。最关键的是,大多数eCO2R半反应的标准电势都接近析氢反应(HER)的标准电势,导致HER 成为水电解质中eCO2R 反应的主要副反应。电化学体系中,相比多芳烃(C2+)产物,CO2分子更易被还原为C1产物。然而,C2+产物比C1产物具有更高的能量密度,在燃料和化学工业中的应用更为广泛,具有更高的经济价值[10]。
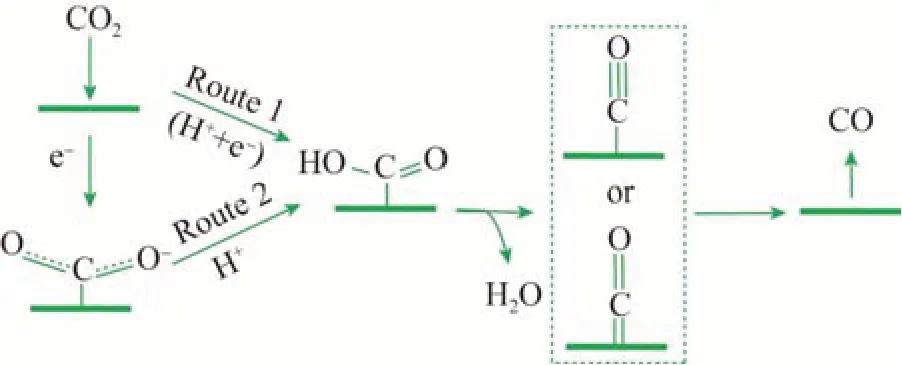
图2 eCO2R制备CO的可能反应机制Fig.2 Possible reaction mechanisms for preparation of CO by eCO2R[5]
2 Cu基催化剂及其性能调控策略
1989年,Hori 等[2]报道了关于eCO2R 反应的一个里程碑式发现:CO2可以在金属Cu 电极上被还原为CO、HCOOH 和碳氢化合物。自此之后,相关研究在世界范围内备受关注,为优异eCO2R催化剂的开发提供机会。在eCO2R的研究过程中,选用过数种金属材料作为催化剂[10]。其中,贵金属Au 和Ag等在eCO2R 过程中对CO 的形成具有选择性,然而,贵金属的高昂成本极大地限制其大规模应用[11-12]。因此,亟需开发储量丰富且具有eCO2R 性能的过渡金属作为替代催化剂。大量研究表明[6],Cu 基催化剂表现出优异的eCO2R 性能和应用前景,可以将CO2还原为碳氢化合物和含氧化合物,呈现极为可观的法拉第效率(FE),但目标产物选择性较差。Cu 基催化剂eCO2R 存在多种还原产物[13-14],如CO,HCOOH,CH3OH 和CH4等4 种最常见 的C1产物;C2H4,C2H5OH 和CH3COOH 等C2产物;C3H8O 和C3H6O 等C3产物。此外,Cu 基催化剂呈现良好的eCO2R运行稳定性,具有较好的应用潜力和前景。
将金属Cu 作为主要原料,通过掺杂、复合和改性等方式对材料的形貌、晶面取向、尺寸大小、表面缺陷、原子组成和表面性质等进行调控而设计不同的Cu 基催化剂,其多种多样的制备方法导致Cu 基催化剂的组成、结构和形貌存在明显差异,最终呈现不同的eCO2R活性和选择性[15]。深入了解各种调控策略对Cu 基催化剂活性和选择性的影响,有助于我们选择合适的合成策略以满足不同的使用环境。
2.1 尺寸设计
通常,纳米催化剂材料因其特殊的形貌结构而具有较高的比表面积,其表面存在的大量边缘/不饱和配位点有利于暴露更多的催化活性位点[16]。随着纳米技术的快速发展,金属材料的尺寸和形貌的相关研究受到了越来越多的关注,以期提高材料eCO2R的性能,其中在纳米尺度调控金属材料的结构对其催化性能有着较为显著的影响。经研究发现,不同的材料尺寸会使材料中局部离子配位数有着较大差异,从而导致对活性组分具有不同的吸附能力。众所周知,单质Cu 作为eCO2R 催化剂面临诸多问题,如过电位大、选择性差、还原时间长和稳定性较差等。基于此,科研人员制备了各种纳米结构的Cu 催化剂[17],包括零维(0D)纳米颗粒、一维(1D)纳米线、二维(2D)层状材料和三维(3D)多孔金属泡沫等,并探讨维度影响活性的各种因素,如尺寸效应、密度效应、应变效应、晶界效应和晶面效应等。
目前,诸如气相沉积、光沉积、喷雾热解、激光烧蚀以及热液途径等技术多被用于制备各类铜纳米颗粒[18-21],经研究表明,与单质铜电极相比,直径5 nm 的铜纳米颗粒能达到比普通材料数倍高的电流密度,但上述技术合成的铜纳米颗粒均大于5 nm,且工艺复杂,所需成本较高,限制了其大范围的应用。因此,对于铜纳米颗粒的合成工艺研究一直受到科研人员的关注。如一种基于使用铜纳米线为前驱体的合成技术,成功获得了负载于碳纳米管的铜纳米颗粒,该样品及其用作铜前驱体的Cu 纳米线负载聚乙烯吡咯烷酮(PVP)的TEM 图像如图3所示。另外,铜颗粒的平均粒径为2~3.8 nm,且相对分散,其HCOOH 和CH3OH 的产量远高于其他较高粒径的样品。由于超细铜纳米颗粒与功能化碳纳米管之间的界面催化位点的存在,使得其也可以eCO2R为C2和C3产物。经过24 h的稳定性测试后,铜纳米颗粒仅表现出弱团聚性,且生产速率稳定,证明该材料具有较好的稳定性[22]。因此,在催化剂设计过程中,需精准把控不同尺寸的材料对催化性能的影响,来选择性合成高催化活性和较好选择性的Cu基催化剂材料。

图3 (a)CuCNT-ImR样品和(b)用作铜前驱体的Cu纳米线的TEM图像(插图为放大图像)Fig.3 TEM images of(a)CuCNT-ImR sample and(b)Cu nanowires used as copper precursors(insert is a magnified image )[22]
2.2 晶面调控
Cu 基催化剂的暴露晶面对于还原产物同样具有重要影响。以晶体单质Cu 为例,Takahashi 等[23]的研究表明,Cu 面心立方晶体的(111)面主要将CO2电催化还原为CH4,而其(100)面则主要生成C2H4。在Cu纳米立方体、Cu纳米八面体和Cu纳米球中,Cu 纳米立方体暴露更多的(100)晶面,其eCO2R 为C2H4的FE 达57%,明显高于暴露(111)晶面的Cu 纳米催化剂。因此,通过调控Cu 基催化剂的暴露晶面改变eCO2R的反应路径,有针对性地促进eCO2R 为目标产物,成为改善催化剂eCO2R 性能的一种有效策略。
对于晶面调控,常使用刻蚀的方法对晶体的表面进行刻蚀,从而暴露出特定的晶面。利用特定的结合剂来选择Cu 纳米颗粒所需要的晶面,对Cu 纳米颗粒的形貌进行选择性刻蚀,如N,N-二甲基甲酰胺可以促进形成Cu 斜方十二面体,油胺可以促进棱角和边缘的刻蚀过程。经选择性刻蚀后形成的纳米颗粒对Cu的(100)面有包覆作用,该具有高能(110)晶面的Cu 斜方十二面体纳米颗粒可以达到远高于其他普通样品的电流密度[24],如图4 所示。由此可知,利用晶面调控进行Cu 基催化剂的设计具有可行性,可以极大地提高其eCO2R的效率,并且表现出一定的产物选择性,使目标产物的调控更精准。
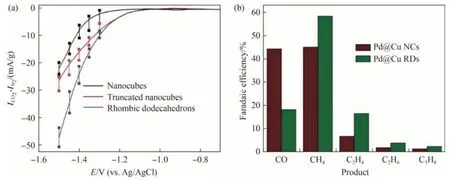
图4 (a)不同形状的铜纳米晶体的CO2电还原电流(根据无CO2电解质中的电流进行校正,纵坐标中为CO2的还原电流密度,为饱和N2环境下析氢反应的电流密度);(b)Cu纳米立方体和Cu菱形十二面体eCO2R的FE值比较(电化学测试在0.25 mol/L K2CO3溶液中进行,扫描速率为20 mV/s)Fig.4 (a)CO2 electroreduction currents corrected for currents in CO2-free solutions for Cu nanocrystals with different shapes(corrected based on current in CO2 free electrolyte, in vertical axis represents reduction current density of CO2,and represents current density of hydrogen evolution reaction in a saturated N2 environment);(b)Comparison of FE values of Cu nanocubes and Cu rhombic dodecahedrons eCO2R(electrochemical tests were performed in a solution containing 0.25 mol/L K2CO3 with a scan rate of 20 mV/s)[24]
2.3 形貌结构调控
理想的催化剂结构意味着材料的形貌需有利于暴露更多的反应活性位点,这就需要对材料的合成进行设计,引入理想的分层、多孔结构,从而增加材料的比表面积。分层结构和多孔结构不仅可以扩大电化学表面积,有利于活性位点的暴露,而且可以使材料拥有协同作用,进而提升材料的催化性能。因此,高孔隙度以及多维层状形貌,同样也是影响eCO2R 催化剂材料催化性能的重要因素。实验表明[25],具有多孔结构的材料不仅有利于促进点还原效率,还能控制合成产物的路径和反应速率,因此对材料孔径的控制也能使其具有一定的选择性。
此外,通过分层结构和孔隙率进行表面调整也可以诱导纳米限域效应[26]。如图5 所示的分层-多孔结构可以增加CO2或还原中间体与催化剂相互作用的可能性[5],从而提高催化剂对CO2的吸附和转化能力。具体而言,大孔(>50 nm)可以促进反应物在每个活性位点之间的传输,从而进行多个电子还原反应。介孔(2~50 nm)和微孔(<2 nm)能够捕获反应中间体和CO2分子,使其在反应通道内停留时间更长,以增加介观通道和纳米通道内的CO2浓度,从而增加CO2转化为复杂C2产物的可能性[23]。此外,根据中间体的分子大小来调整孔隙和通道的直径也是一种有效调控方法。Lv 等[27]制备了一种多孔Cu 电催化剂,孔径分布为100~200 nm。增大的表面积和多孔通道显著改善了电解过程中气体通过电极-电解质界面的转移过程。该催化剂在-0.67 V(vs.RHE)电位表现出653 mA/cm2的eCO2R 电流密度,且C2+的FE 可达到约62%。进一步的研究表明,这些多孔通道内的局部pH 与体电解质的pH 有本质上的不同,从而控制了eCO2R 的反应途径。但在稳定性测试中,该催化剂在200 mA/cm2的电流密度下仅能稳定运行2 h。目前已有较多研究利用表面活性剂和温度控制制备核壳结构的催化剂材料,且已证明了该结构较好的稳定性,在较长时间的电催化测试中可保持良好活性[28]。因此,对于Cu 基催化材料的设计合成,不仅要兼具材料形貌对催化活性以及选择性的影响,同时也要赋予材料相对稳定的结构来保持其在eCO2R过程中的稳定运行。

图5 具有选择性的分层-多孔结构示意图Fig.5 Schematic diagram of a hierarchical-porous structure with selectivity[5]
2.4 单原子材料
传统均相催化剂和分离多相催化剂有着较大的差别,虽然传统均相催化剂具有选择性高、副反应少且催化活性高等特点,但催化剂与反应物和产物的分离以及回收问题一直是阻碍其广泛应用的重要原因。而催化效率较低的分离多相催化剂却更有利于回收循环。因此,将催化剂负载化,结合均相催化剂与分离多相催化剂的优点,可以有效地提升催化剂效率。经研究得知,负载于衬底的金属组分颗粒粒径足够小,使得表面原子数占比大幅提升,有利于提高催化效率[29]。因此将金属以单原子的形式负载于衬底上时,构成单原子催化剂,能极大地提升金属原子的利用率,促进活性位点的增加[14]。同时具备均相催化剂与多相催化剂的高效率与易分离等优势的单原子催化剂,不仅极大减少了原材料的使用量,单原子金属带来的尺寸效应以及与衬底之间的相互协同作用使其拥有较为独特的电子结构,从而成为一种越来越受关注的催化剂类型。金属铜单原子催化剂有着足以比拟贵金属催化剂的催化性能,解决了生产成本这一关键瓶颈[27]。
在衬底上负载金属Cu 单原子不仅可以极大降低金属Cu 的用量,还可以保持甚至更优于普通Cu电极的性能。Song 等[30]设计了2 种具有不同不对称原子界面的Cu 单原子催化剂,利用密度泛函理论(DFT)计算不同催化剂模型的吸附能并进行比较(图6)。研究表明,催化剂CuN3O/C 具有很高的eCO2R选择性,在-0.8 V(vs.RHE)电位时eCO2R为CO 的FE 达96%,而CuCO3/C 对CO 的生产选择性较差,在-0.5 V(vs.RHE)时的FE 值最高只有20.0%。DFT 结果表明,在决定CO 解吸速率的步骤中,CuN3O 位点需要比CuCO3更低的吉布斯自由能,从而使其拥有更高效的催化活性。
由此可以看出,Cu 单原子催化剂可以展现出单原子材料在eCO2R中的独特优势[16]。然而,单原子催化剂仍然面临活性位点有限和稳定性较差的问题。而且单原子催化剂的制备通常需要苛刻的条件,如高温热解或强酸蚀刻。未来,Cu 单原子在支撑物上的负载量以及催化过程中的活性位点等问题是提升单原子催化剂性能的研究重点。
2.5 双金属催化剂
随着对eCO2R的深入研究,尽管单原子催化剂表现出优良的催化活性和选择性,但由于单原子催化剂较简单的活性中心结构限制了其活性的进一步提升。因此,利用不同原子之间的协同作用来调节催化剂的反应活性这一策略被提出,并逐渐受到了较广泛的关注。Cu 作为一种至关重要的eCO2R 催化材料同样也被用来制备不同的双金属催化剂,利用两种金属之间对中间产物的不同键合作用和不同的金属位点相互作用进而提升催化剂的内在活性,且双金属异质材料往往具有相互串联的结构,有助于催化剂的稳定性[31-32]。如已报道的Fe/Cu 双金属催化剂对CO 具有将近98%的FE值,并保持较长时间的稳定性[33-34]。此外,双金属催化剂不仅可以引入协同作用,其催化产物的比例也可根据对双金属催化剂中不同原子的比例进行调控。如Chen等[35]合成了含3.2%Cu的C-Cu-Co-ZIFs 催化剂,经测试得,在-0.55 V(vs.RHE)的电压下,其对CO 与H2的FE 值分别为51%与37%。在-0.7 V(vs.RHE)的电压下,CO 的电流密度可达8.3 mA/cm2。另外,具有相似结合亲和力的不同金属原子也被设计为双金属催化剂来催化CO2还原。如Cheng等[36]合成了MOF支撑的Ni/Cu双位点催化剂。经表征测试得知,Ni/Cu 相邻位点之间会诱导电子效应,使催化剂发生电荷再分配,且Ni/Cu 活性位点与相邻N4Ni/CuN4基团之间的协同效应增加了材料的导电性以及与活性中间体的相互作用。在-0.79 V(vs.RHE)下对CO 的FE 值高达99.2%,在-0.39~-1.09 V(vs.RHE)对CO 的FE 值达到了95%。由此可见,构建双金属催化剂是调控Cu 基催化剂性能的一种非常有效的策略。
3 Cu基催化剂的还原产物
通过电化学还原的方法,可使CO2在Cu 基催化剂表面得到电子后转化为其他化合物,但eCO2R反应复杂、中间反应物多且还原产物多,如多种C1和C2+产物。由于Cu 表面对CO2的还原具有选择性,不同的反应环境、催化剂和电位大小都会对还原产物产生一定的影响。利用不同条件产生的不同产物,对其进行分类收集,是提高产物利用率的有效措施。下面详细阐述了Cu 基催化剂eCO2R 的各种还原产物以及不同碳产物的可能路径与机理。
3.1 C1产物
Cu 基催化剂eCO2R 为C1的产物主要是CO,CH4和CH3OH 等。通常电催化还原CO2形成CO 的过程主要是通过利用*OCHO 中间体的过程,但这一过程往往伴随着析氢副反应的影响,要想减少析氢副反应,则要求中间体和催化剂的结合更紧密。此外,气态烃类在铜箔上的FE 相当高,在电流密度为5 mA/cm2[电位E=-1.8 V(vs.RHE)]时,烃类产物的FE 最高可达60%。当CO2吸附在Cu 电极表面的活性位点时,在-0.8 V(vs.RHE)的电位接受电子和质子,结合成变形H-C=O结构,随后不断地还原和接受质子和电子,逐渐转化为CH4或者CH3OH,具体过程如图7所示[37]。
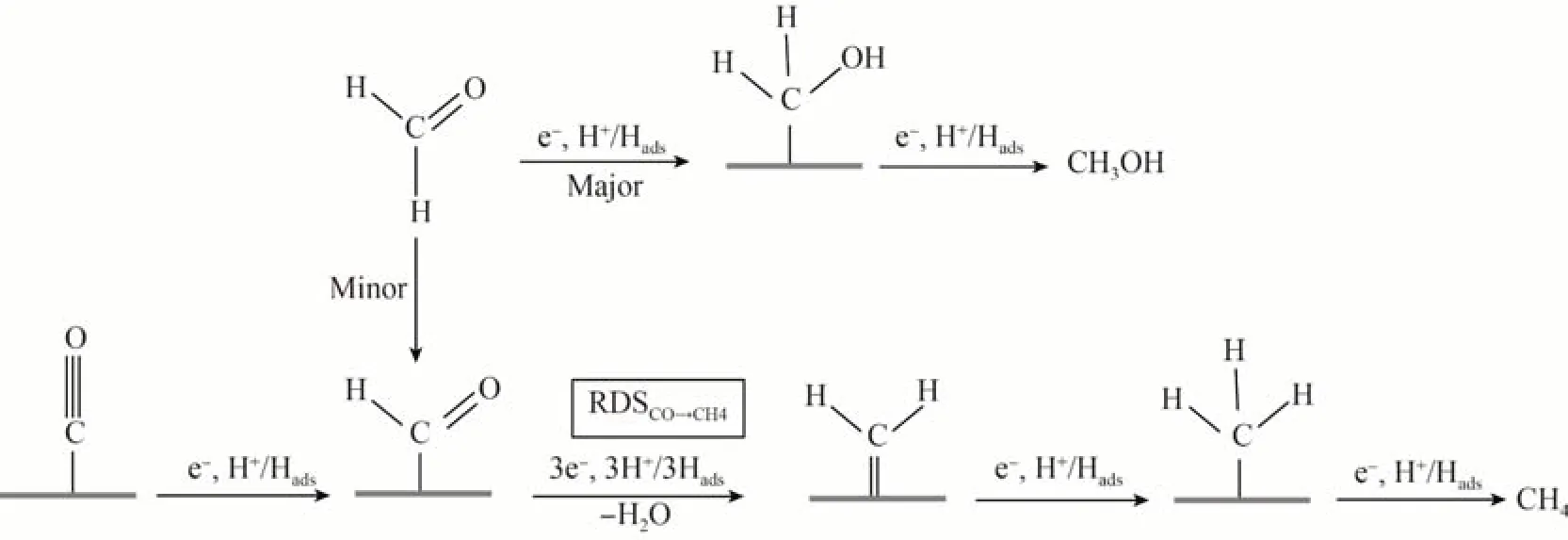
图7 C1产物形成机理Fig.7 Mechanism of C1 product formation[37]
当然,对于Cu 基催化剂的不同的调控措施也会影响C1产物的形成。如Huan 等[38]设计的多孔树枝状铜基材料,其在水环境下甲酸盐的FE 值可达87%。Takashima 等[39]合成的高Pd/Cu 比合金,其eCO2R 产物主要是HCOOH,同时该调控措施也会改善催化剂的稳定性,在长期电解还原电流的运行下仍能保持稳定,减少了单一金属催化剂因CO失活的可能。此外,有报道显示,通过非均匀气相沉积制备的含表面氧化层的OFn-Cu 纳米颗粒提高了CH4最大形成电流,同时也证明O 位点可以增强催化反应活性[40]。而借助脉冲偏压等外力的方式也被应用于对反应的路径进行调控,如Strain 等[41]利用该方法减少铜衍生氧化物催化剂的析氢副反应,使其生成CO 的量更多。因此,了解不同调控策略对反应路径以及C1产物的产生种类的影响,可以使后续研究根据需求选择合适的方法以促进C1产物的生成。
3.2 C2产物
Cu 基催化剂eCO2R 过程中,生成的C2产物有很大的利用价值,C2产物在工业发展中具有举足轻重的作用。C2H4和C2H5OH 等C2产物作为原料或者能源的使用已经有了较长时间,具备相对完整的工业体系,比如被称为工业血液的C2H2。此外,Cu基催化剂对CO2的强结合力使得其更倾向形成C2产物。基于以上两点,发展利用Cu 基催化剂eCO2R生产C2产物具有显著的研究意义。因此,在进行eCO2R过程中,催化剂的使用需尽量去贴近C2的反应过程。合成C2产品是一个较为复杂的过程,经过大量的实验和理论研究,科研人员已经得出一些可能的反应机制[5]。C2产物的形成可以通过不同的C1中间体二聚或偶联进一步还原为C2产物。图8 阐明了Cu 基催化剂eCO2R 合成7 种C2产物的3种公认的形成路线,主要产物包括C2H4,C2H5OH和n-C3H7OH,次要产物包括(FE 在1%~10%之间)C2H6和CH3COO-,以及微量产物C2H2O2和(CH2OH)2(FE 小于1%)[5]。几种平行反应途径的共存导致eCO2R 反应形成各种可能的还原产物。C2H6和CH3COO-的形成共享路径1,其中C2H6通过*CH3的二聚化(路径1B)转化,而CH3COO-的形成遵循CO插入机制(路径1A)。对于整个路线1,*CH2是C2产物形成的关键中间体,与CH4的形成相竞争,被认为是选择性决定中间体(SDI)。对于路线2,C2H2O2和(CH2OH)2均源于*CHO 中间体,其次是CO 插入机制。然而,*CHO 也是通过质子耦合电子转移(PCET)过程形成CH4的共同前体。由于所需能垒低,该路线有利于CH4产物的形成而非C2产物。对于路线3,通过*CO 二聚反应的C-C 耦合步骤被视为整个路线的速率决定步骤(RDS)。经过逐步质子化和电子转移,形成的*CH2CHO 作为形成C2H4和C2H5OH 的SDI。相比之下,由*CH2CHO形成C2H5OH 的能垒[约0.2 V(vs.RHE),路线3B]高于*CH2CHO 还原为C2H4(路线3A)[42]。这也是在实验中大块Cu 表面上eCO2R 对C2H4的选择性高于C2H5OH 的原因[43]。在研究过程中发现,晶界这种非平衡结构更倾向C2产物[44]。通过物理气相沉积法制备的石墨烯承载铜纳米颗粒可以观察到大量晶界结构,并在eCO2R 的过程中对C2H5OH 和乙酸盐展现出高选择性。同时,局部酸碱度升高也被证实会促进C2产物形成,Yang 等[45]利用溅射铜的方法获得明确孔隙率的电极,在-1.3 V(vs.RHE)时直径为30 nm、深度为40 nm 的孔主要产生乙烯,更深的孔则会生成饱和度更高的乙烷,更宽的孔产物主要为C1。作者认为原因在于催化剂不同尺寸的孔影响局部pH值,由于阴极产生OH-使质子反应受阻。还可以利用电解原理,解构Cu 纳米立方颗粒密集集合结构[25],增加C1中间体二聚或偶联的机会,以此来提高C2产物的生成速率。以导电碳材料为载体合成枝化CuO 电极也是利用上述方式,增大表面积、提高连接以此快速消耗质子,从而增加局部的酸碱度。在了解了反应的机理和催化剂的制备方案后不难看出,增加接触和提升耦合过程,有利于C2产物的生成。
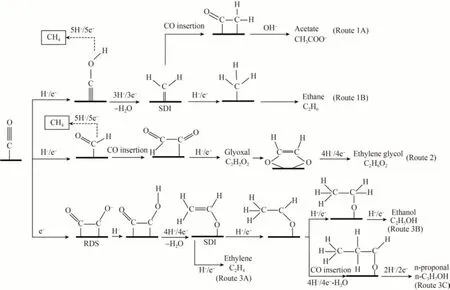
图8 低过电位下Cu表面的*CO可能演变为C2的途径Fig.8 Possible pathway of *CO on Cu surface evolve into C2 at low overpotentials[5]
3.3 多碳产物
多碳产物主要为C2+类别,其主要特点为碳链较长。高碳链意味着有更多的能量,同时也造成了其更难合成的事实。C2+产品较高的市场赋值得到了化工产业的广泛关注。目前,随着全球市场能源结构的变化,能源市场的动荡使得多碳产物的生产有重要的现实意义。但是在目前现有的技术上,C2+产物的生产效率仍较低,大多数生成的C2+产物的分电流密度均低于1 mA/cm2。Cu 作为唯一对*CO 具有负吸附能,且对*H 有正吸附能的金属,可以使*CO 的激活和转化达到平衡,这也是合成多碳产物的关键[25]。图9 展示了eCO2R 在Cu 催化剂上生成C2+产物的可能路径。显然,这是一个高度复杂的过程,eCO2R 过程中,CO2首先通过*COOH 中间体被还原为吸附的*CO,而*CO 物种是形成C2+产物的关键中间体[15]。同时,C2+还原产物的最后分布也和*CO 中间体的后续转换息息相关。最关键的是,C-C偶联是C2+产物形成的核心步骤。目前被广泛接受的观点是,C-C 耦合机制是CO 的直接二聚化[10],该结果得到了控制实验、光学计算和原位光谱等研究的证实。
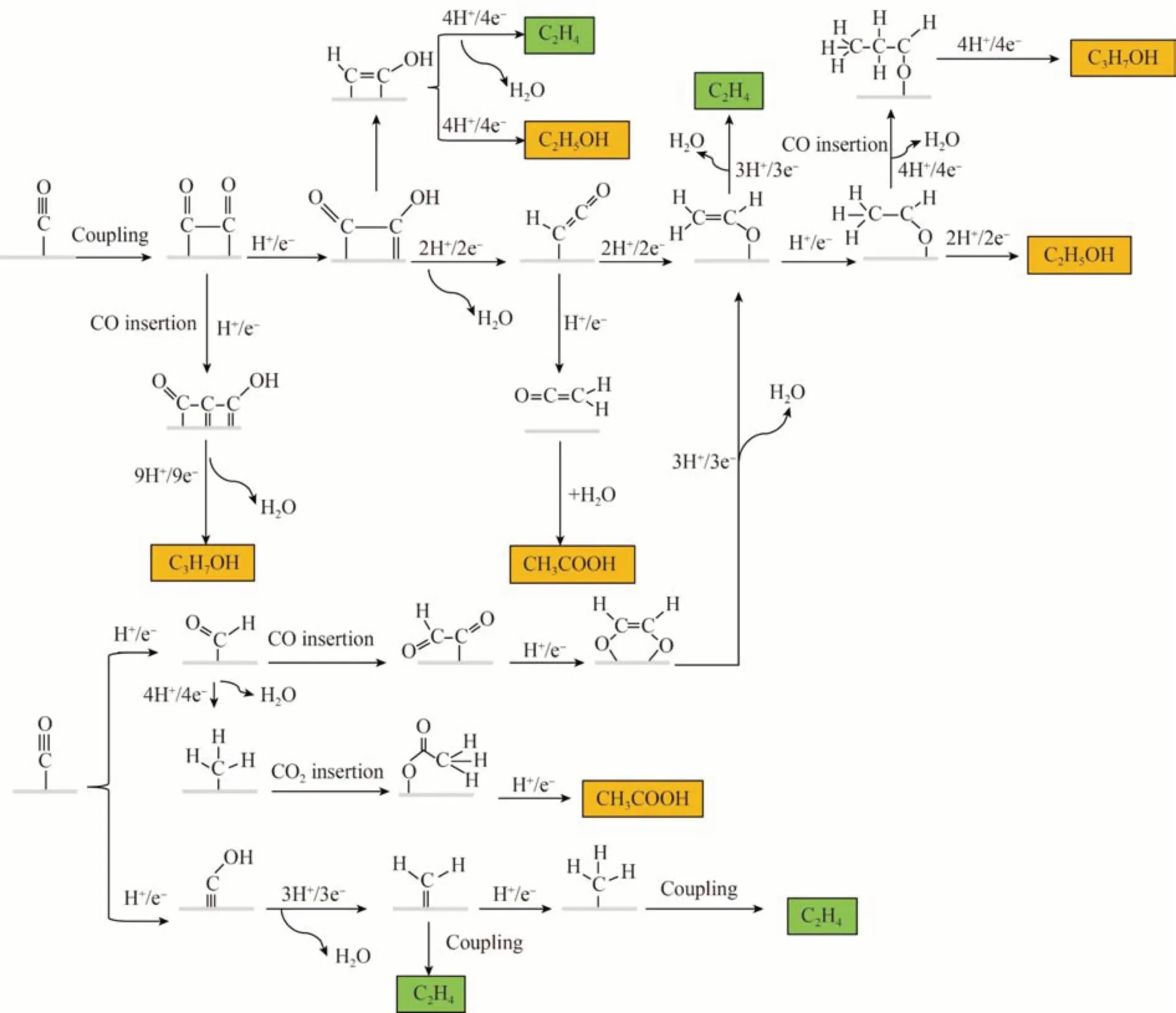
图9 部分多碳产物的合成路径Fig.9 Synthesis pathways for some of multi-carbon products[10]
含水环境中,线性CO2转化为弯曲的化学吸附CO2,作为CO2回收并发生还原反应的第一步。吸附后,一般的CO2初始反应被认为是CO2-物质的生成,由于其寿命短,难以检测和表征,因此提出了扫描电化学显微技术(SECM)的尖端生成/底物收集(TG/SC)模式,以成功捕获检测信号。特别是通过将实验数据与理论模型进行拟合,可以检测CO2的二聚速率(6.0×108L/mol)。
目前来说,利用疏水层的高分子和尖端的Cu表面支晶结构构造出的材料的接触角可达到153°[10]。这样可以增加CO2的吸附量,从而提高eCO2R 产物的FE,并且增加更多的C2+产物。另外,多孔有机笼改性的Cu 纳米型催化剂上,C2+产物的最佳FE 可达到76.1%,在-0.9 V时,电流密度为1.7 mA/cm2。此外,该策略也适用于多孔无机碳材料调制的Cu 基催化剂,与原始铜或碳黑修饰的电极相比,涂有多孔碳气凝胶的铜箔由于碳气凝胶中的疏水通道促进了CO2分子向铜-碳界面的扩散而促进C2+产物的形成[46]。Cheng 等[47]合成了核壳纳米立方形的Cu2O@K-PBA,使产物的选择性向多碳方向进行,且该结构可以把活性物质限制在一定区域内,增加C-C 耦合概率,也可以保护活性位点,增强材料的稳定性。也有报道制备氧化亚铜膜衍生电极[43],主要在基体上通过电沉积或水热合成的方法制备,制备成的电极通常有粗糙的氧化还原表面层,可以抑制CH4的生成,提高C2+产物的量。由此可见,无论是构造立体枝晶、核壳结构或者粗糙氧化表面,想要促进C2+的选择性的首要目标就是增加催化剂和原料的接触,同时注意保护活性位点,以提高催化剂本身的稳定性,这样才更加有利于碳催化过程向更多碳数、更长碳链的方向进行。
4 总结与展望
本文综述了Cu 基催化剂的各种调控策略及其相关影响因素,并系统总结了Cu 基催化剂eCO2R的多种催化产物以及eCO2R 形成不同碳产物的可能路径与机理。目前看来,CO2由于本身结构和键型的原因,在热力学上是较为稳定的分子,在动力学上,通过Cu 基催化剂eCO2R 的过程包含较多的中间过程且反应较为复杂。目前,利用合理的合成策略设计Cu 基催化剂还原C1,C2和C2+的过程已经取得巨大进展,有巨大的发展前景。Cu 基催化剂的尺寸、晶面、结构和成分都对其本身的催化活性、稳定性以及产物的选择性有一定的影响。在设计催化剂的过程当中,需倾向于暴露更多活性位点,促进多碳产品的形成,最大限度提高原子利用效率,抑制HER的发生。
目前对于Cu 基催化剂eCO2R 的详细反应机制仍不清楚,需要从实验和理论两方面进一步深入研究。同时,由于Cu 基催化剂eCO2R 生成C2和C3产物的过电位较高,且对特定产物的选择性也远不能满足需求,因此整体催化性能低下。在提升催化性能的策略方面,不同的策略在提高eCO2R性能方面的工作原理通常是不明确的,因此不同策略的作用在彼此之间可能有一些重合。另一方面,电解质对催化性能的影响是非常模糊的,因为很难检测到电极表面附近的离子行为。综上,未来的研究重点可能倾向于以下几个方面:
1)eCO2R 的反应途径/机制具有多步质子和电子转移的特点,其中涉及多种产物相关的各种反应中间体。为了揭示eCO2R的复杂过程,需将先进的操作性表征技术与理论计算相结合,进一步理清反应的复杂机制。
2)揭示每一种催化调控策略在Cu 基催化剂上的具体作用,并通过原位表征工具确定eCO2R的关键中间体,正受到越来越多的关注。先进的原位技术,如原位扫描隧道显微镜、原位透射电子显微镜、原位扫描电子显微镜、原位X 射线衍射和原位X 射线光电子能谱,可以记录eCO2R 过程中CO2的转化过程,有助于揭示Cu 基催化剂的真实情况。此外,一些原位技术,如原位傅里叶变换红外光谱法、原位拉曼光谱法和原位质谱法等,其在检测eCO2R 的中间产物方面很敏感,这将为揭示不同eCO2R 产物的基本途径提供一种有效策略。而如何利用以上先进的表征手段来探究各中间体在反应过程的作用是促进催化剂性能提升的关键。
