HIV-1 新型重组毒株近似全长基因组结构鉴定分析
2024-01-12路新利王莹莹
路新利 刘 勇 李 岩 王莹莹 安 宁 刘 萌
(1.河北省疾病预防控制中心艾滋病防治所,河北 石家庄 050021;2.河北省邱县中心医院,河北 邯郸 057450)
1981年,美国报道了全球首例获得性免疫缺陷综合征(acquired immune deficiency syndrome,AIDS)病例[1]。40年来,人类免疫缺陷病毒(human immunodeficiency virus,HIV)广泛传播,已成为全球性的重大公共卫生问题。截至2020年年底,全球有3 770万例HIV感染/AIDS患者[2]。HIV主要分为HIV-1和HIV-2这2种类型,HIV-1是造成全球大流行的主要病原毒株,具有极强的复制、变异和重组能力,遗传性复杂、多样,是AIDS防治和疫苗研制的主要障碍[3]。本研究对河北省3例经男男同性性接触感染HIV-1的男男同性恋(men who have sex with men,MSM)病例的血浆样本进行HIV-1近似全长基因组序列(near full-length genome,NFLG)分析,确定其重组模式,为研究HIV-1遗传多样性提供数据支持。
1 材料和方法
1.1 研究对象
3例患者(编号分别为150、110和171)均为MSM,2019年于河北省艾滋病确证中心实验室确证为HIV-1抗体阳性,均通过不安全的同性性行为感染HIV-1。3人均已婚或同居,年龄分别为43、52和41岁,血液中HIV-1含量分别为5.45×105、3.02×105和2.54×104拷贝/mL,CD4细胞数分别为263、479和135个/μL。所有病例暴露前均未服用暴露前预防用药,未进行过抗HIV-1治疗。在对这3例病例进行耐药基因型检测时,初步分析其基因亚型分别为B/C、B/C和01_AE/B。本研究经河北省疾病预防控制中心伦理委员会审核通过[IRB(S)-2020-002]。
1.2 方法
1.2.1 HIV-1 RNA提取和目的基因组扩增
采集患者静脉血样本,离心分离血浆,-80 ℃保存待检。参照德国Qiagen公司QIAamp Viral RNA Mini Kit说明书,从140 μL血浆样本中提取HIV-1 RNA。采用日本TaKaRa公司Prime Script One Step RT-PCR Kit和LA Taq,参照张菲等[4]的方法设计引物进行NFLG(HXB2:649-9599)扩增,扩增产物送北京博迈德基因测序公司进行测序。
1.2.2 序列拼接比对和系统发育分析
用CExpress 9.1软件将每个患者样本的所有原始反应序列拼接成1条完整的NFLG,使用HIVAlign在线软件进行基因序列比对(https://www.hiv.lanl.gov/content/sequence/VIRALIGN/viralign.html),然后通过BioEdit 7.0软件进行手动编辑和校对。采用MEGA 7.0软件,基于邻近法和Kimura 2-parameter model构建neighborjoining进化树,以Bootstrap值≥70%为有意义。不同亚型参考株(A-D、H-K、O、CRF)的NFLG下载自HIV Databases数据库(https://www.hiv.lanl.gov/)。
1.2.3 NFLG基因组镶嵌结构分析
首先用HIV Databases数据库中的jpHMM在线软件初步分析3条NFLG的基因重组图谱。然后用SimPlot v3.5.1软件进一步分析NFLG与参考序列的相似性和重组模式,确认NFLG的镶嵌结构和基因重组断点。
2 结果
2.1 系统发育树分析结果
经过核酸提取、扩增和序列测定,获得3条HIV-1 NFLG(GenBank序列编码:ON959789-ON959791)。NFLG 150、NFLG 110和NFLG 171长度分别为8 966、8 740和8 994 bp。基于这3条NFLG与参考序列构建neighbor-joining进化树,在进化树上,病例110、150和171的NFLG与其他各参考亚型序列距离较远,分别形成了单独的分支,其中110和150与CRF07_BC具有共同的根,见图1。说明这3例患者的NFLG可能是新型重组毒株,需要通过基因重组断点分析进行确认。

图1 基于NFLG的neighbor-joining进化树
2.2 NFLG jpHMM在线软件分析结果
基于HXB2参考株的基因位点,发现与参考株CRF07_BC的基因组图谱相比,病例110和150的jpHMM基因组图谱右边的env区基因重组断点基本一致,但是在gag-pol区存在差异,病例150的jpHMM基因组图谱分别在gag区1 270~1 411位点和pol的3 011~3 311位点缺少2个C和B的基因重组断点,初步判定其可能为有C亚型和B亚型参与重组的新型重组毒株。病例110的jpHMM基因组图谱在gag区1 270~1 411位点没有重组断点,并且在pol区的3 108±119位点有基因缺失,说明该病例存在多种亚型毒株重组的可能。jpHMM基因组图谱显示病例171的NFLG与CRF01_AE一致。见图2。

图2 近似全长基因组的jpHMM重组分析
2.3 近似全长基因组基因相似性分析
采用SimPlot v3.5.1软件对这3条NFLG和不同亚型的标准参考毒株进行基因相似性分析,并确定基因重组断点位置。结果显示,NFLG 150是由CRF01_AE和CRF07_BC这2个循环重组形成的新型二代重组毒株,该序列的基因重组模式是2个CRF01_AE亚型片段分别插入到CRF07_BC亚型基因组骨架中,在BootScan图谱上,插入重组位点1个在gag区的740~800位点,另1个在env区的8 320~8 400位点,见图3。然而,由于插入的2个CRF01_AE片段均较短,分别约为60和80 bp,如果进行基因亚区进化树分析难度较大,极有可能会与CRF07_BC参考序列集聚成簇,因此本研究未对其进行基因亚区进化分析。病例110和171的NFLG BootScan分析图谱显示,这2条NFLG分别为CRF07_BC亚型和CRF01_AE亚型,不是新型重组毒株,见图4。

图3 病例150的NFLG BootScan分析图谱
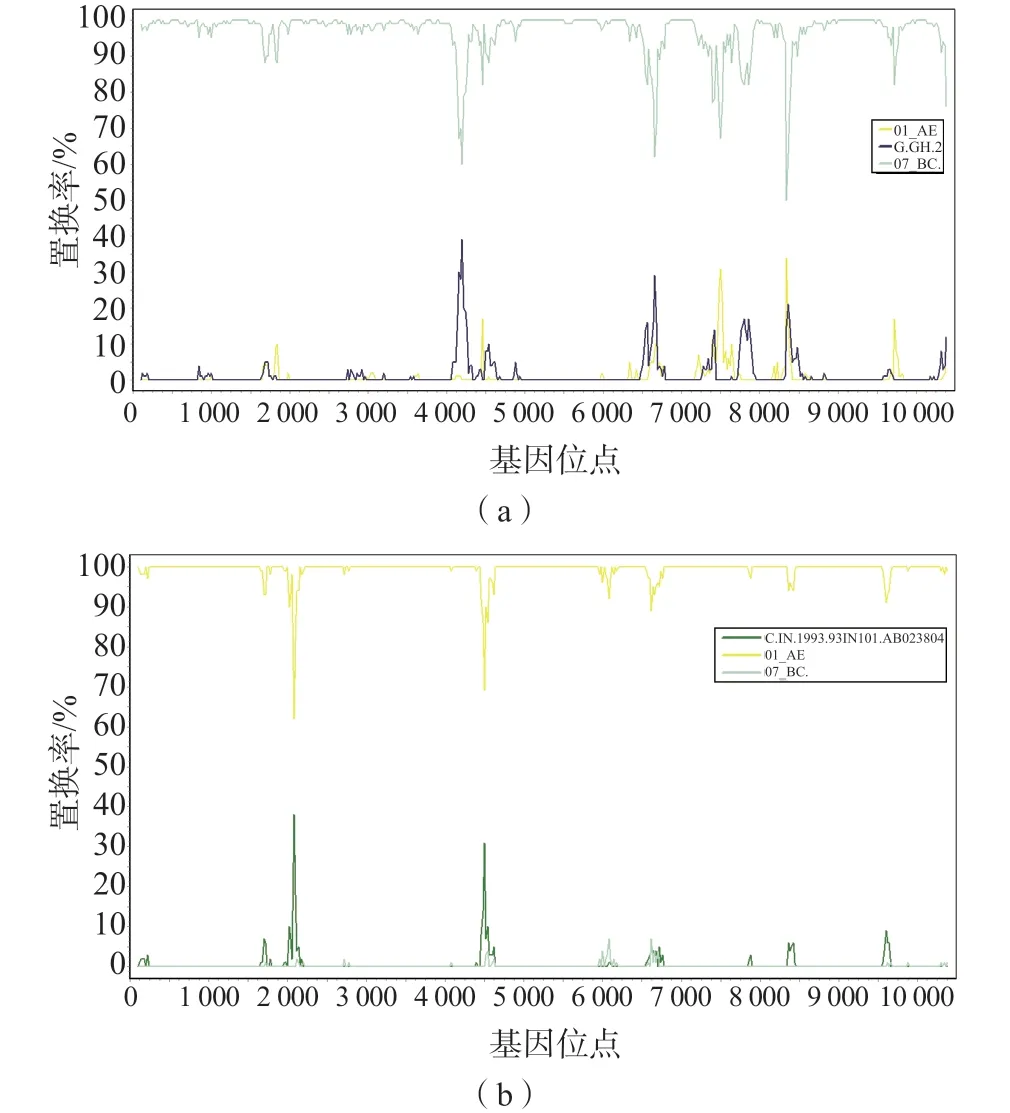
图4 病例110和171的NFLG BootScan分析图谱
3 讨论
斯坦福大学HIV公共数据库(https://www.hiv.lanl.gov/components/sequence/HIV/ search/search.html)显示,目前全球已有118种CRF和大量的独特型重组毒株(unique recombinant form,URF)被发现。我国国家分子流行病学调查结果显示,随着传播途径的转变,CRF和URF数量快速增多,HIV-1亚型毒株的分布也发生了明显变化[5]。当前,性传播已成为我国HIV最主要的传播途径,CRF07_BC和CRF01_AE是我国最主要的流行毒株[6],它们的共流行和双重感染为二代重组创造了条件。在上世纪90年代,CRF01_AE由泰国通过异性性传播传入我国,最早在我国云南、广西等地区异性传播人群和吸毒人群中流行[7-8]。CRF07_BC是在我国云南地区吸毒人群中发现的重组毒株[9],后来随毒品网络传至四川、新疆等地区。随着传播途径由血液传播、注射吸毒传播向性接触传播转变,CRF01_AE和CRF07_BC快速向其他人群蔓延,特别是MSM人群。
2005年之前,河北省HIV-1主要以血液传播为主,B亚型是最主要的流行毒株[10],随着传播方式向性传播,特别是MSM传播转变,2005年以后,CRF01_AE逐渐成为河北省最主要的流行毒株,B亚型居第2位[11]。相关研究结果表明,随着CRF01_AE的广泛传播,其在我国已经形成7个流行传播簇[12]。CRF07_BC的传播主要经历了2个指数增长阶段[9],分别由07BC_O和07BC_N驱动。07BC_O是原始CRF07_BC传播簇,在注射吸毒者和异性恋人群中传播,主要分布在我国西南和西北地区;07BC_N是一个新的群体,主要在我国北方的MSM中发现,随着时间的推移呈递增趋势,逐渐取代了07BC_O。与广西、云南等地区流行的经异性性接触感染和吸毒人群流行簇不同,河北省CRF01_AE传播簇主要以MSM流行传播簇为主[11],CRF07_BC的主要传播簇是07BC_N。特别是这些流行簇与北京、辽宁等相邻地区的MSM人群紧密相关,聚集成簇[13],具有较大的传播风险。
有研究发现,一个地区同时流行多种毒株是造成双重感染、多重感染和新型基因重组的主要原因,如广西地区的CRF01_AE/CRF07_BC/CRF08_BC三重重组[14],贵州、辽宁地区的CRF01_AE/CRF07_BC双重重组[15-16]。CRF01_AE和B亚型作为主要流行毒株,经过在河北省省内长时间的流行,已经形成大量具有不同镶嵌结构的新型CRF01_AE/B重组体[11],有的已广泛流行,转化成为CRF,如发现于保定地区的CRF103_01B毒株[17]。然而,CRF01_AE/CRF07_BC二代重组鲜有报道。从2021年开始,连续在MSM人群中发现2个具有不同重组断点的CRF01_AE/CRF07_BC重组体[18-19],提示随着CRF07_BC在河北省新报告病例中所占比例上升到第2位,有其参与重组的重组毒株将会越来越多。
本研究确定了以CRF07_BC 为骨架的CRF01_AE/CRF07_BC重组毒株,重组断点显示,2个小的CRF07_BC基因片段分别在gag区和env区插入到CRF07_BC基因组骨架中,明显不同于2021年报道的二代重组模式[18-19],再次确认了河北省HIV-1遗传基因的多样性和流行情况的严峻。另外,河北省发现的CRF01_AE/CRF07_BC二代重组毒株均来自MSM人群,我国已在MSM人群中发现6种CRF[17]。目前,河北省HIV传播仍处于上升期,报告AIDS病例以青壮年为主,性活动活跃,近10年来性传播持续成为主要传播途径,占96.9%,其中MSM者占64.2%[20]。在河北省已发现的HIV-1亚型均已在MSM人群中流行,并且CRF01_AE和CRF07_BC已分别在以MSM为主的性传播人群中形成6个和1个大传播簇,与邻省形成紧密传播关系[11,13],这为新型重组毒株的形成和传播创造了条件。自2013年CRF07_BC超越B亚型成为河北省第2位流行毒株以来,河北省关于HIV-1全基因组重组结构的研究缺失,提示应加强对性传播,特别是MSM人群HIV-1基因多样性和传播特征的研究,及时掌握河北省基因重组毒株流行情况,干预该人群传播簇的形成,阻止其向外传播和形成新的CRF。
