DNA甲基化对水稻花药培养过程中黄化的影响
2024-01-10龚志云
王 笑, 刘 凯, 孙 尚, 薛 超, 龚志云
(扬州大学农学院/ 江苏省作物基因组学和分子育种重点实验室/ 植物功能基因组学教育部重点实验室/江苏省作物遗传生理重点实验室/ 江苏省粮食作物现代产业技术协同创新中心, 江苏扬州 225009)
花药培养是常用的植物组织培养技术之一,能产生具有特定亲本特性的单倍体,然后通过自然或人工加倍获得纯合子二倍体,大大加快育种速度,因而广泛应用于培育各种水稻栽培品种[1]。但水稻花药培养中常产生大量黄化苗,有时高达80%~90%[2]。这已成为花药培养在水稻育种中进一步发展和应用的重大障碍之一。基因突变曾被认为是导致幼苗黄化的主要原因,但其发生频率如此之高并不符合基因突变的规律。因此,急需对水稻花药培养中高频黄化的分子和遗传调控机制进行深入研究[3]。
愈伤组织的诱导和分化是水稻花药培养的2个重要过程。脱分化及营养、光温不足等不利因素均可能阻碍愈伤组织的形成。这些因素会刺激表观遗传调控的变化,因此黄化苗的出现可能与表观遗传调控有关[4]。尽管水稻基因组已完全测序,但关于水稻花药培养表观遗传学的相关报道却很少[5-6]。在植物生长过程中,重金属、干旱、盐、高温等诸多不利因素经常阻碍植株的正常生长。生物通常进化出相应的调节机制(基因转录、翻译调控或表观遗传学)来抵御这些不利因素[7-8]。DNA甲基化是一种不改变DNA序列的保守表观遗传学机制,由DNA甲基转移酶催化基因组CpG二核苷酸中胞嘧啶第5碳位的甲基发生共价结合反应而产生,通过改变染色质结构、DNA构象和DNA稳定性导致基因表达水平的改变[9], 在植物基因组稳定性、环境响应和发育调控中具有重要意义[10]。以往研究[11]表明,水稻在组织培养或再生过程中易获得或失去DNA甲基化,影响其正常生长发育。
本研究通过亚硫酸盐测序(BS-Seq)和RNA测序(RNA-Seq)技术,分别对水稻花药培养中产生的绿苗和黄化苗进行转录组和甲基化组分析,确定它们之间的差异性; 利用SPAD叶绿素仪和分光光度计检测黄化苗叶绿素含量,利用透射电镜成像技术观察叶绿体形态的变化; 通过对绿苗和黄化苗的差异DNA甲基化基因以及转录组水平中差异表达基因的分析,探究花药培养过程中高频黄化苗的产生可能与DNA甲基化的影响机制。
1 材料与方法
1.1 试验材料
供试品种为粳稻品种日本晴。用湿润的纱布包裹水稻幼穗,放入4 ℃冰箱预处理7 d。经消毒、冲洗等处理后,将合适的颖花花药均匀地撒在花药培养基表面,置于(25±1) ℃的暗环境中培养,培养3~4周后开始观察花药对诱导的反应。然后将愈伤组织转移到再生培养基上,在(25±1) ℃的人工光照下培养。最后将再生的绿苗和黄化苗转移到生根培养基中形成根系。
1.2 光合色素含量的测定
取生长14 d的绿苗和黄化苗用于光合色素含量的测定。利用SPAD-502型叶绿素仪(日本柯尼卡美能达公司)测定叶片上、中、下部的透光系数,重复3次,计算平均SPAD值。利用紫外/可见分光光度计(Lambda650型,美国铂金埃尔默公司)测定5张叶片不同部位的色素含量。
1.3 叶绿体透射电镜观察
采集生长14 d的绿苗和黄化苗叶片,切成约1 mm×1 mm的小片。样品用25%戊二醛磷酸盐缓冲液(pH 7.4)在4 ℃下预处理4 h, 用0.1 mol·l-1PBS清洗3次, 每次15 min。接着用锇酸固定 4 h, 再用0.1 mol·l-1PBS清洗3次, 每次15 min。然后用梯度乙醇(分别为50%、70%、80%、90%、95%、100%)溶液、100%丙醇和100%丙酮与无水Na2SO4逐级脱水。丙醇与树脂1∶1混合浸泡1 h, 丙酮与树脂1∶2浸泡2 h, 纯树脂浸泡2 h。用LR White树脂浸渍, 60 ℃聚合2 d。用Lecia EM uc6超显微切片仪(德国徕卡公司)制备超薄切片,超薄切片染色正常,并用Tecnai Spirit (120 kV)透射电镜(荷兰飞利浦公司)观察叶绿体结构。
1.4 DNA提取和BS-seq文库构建
使用DNeasy Plant Mini (Qiagen 69104, 德国凯杰公司)从绿苗和黄化苗的叶片中提取总基因组DNA (gDNA)。使用琼脂糖凝胶电泳和紫外分光光度仪评估基因组DNA质量,使用Bioruptor (比利时Diagenode公司)将gDNA切成100~500 bp的片段。在末端修复、腺苷酸化和接头序列连接(以防止亚硫酸盐转化)后,以ZYMO甲基化试剂盒(美国ZYMO Research公司)用亚硫酸氢盐处理DNA片段。处理后DNA在自旋柱上纯化,用于制备测序文库。在这一过程中,亚硫酸盐处理的单链DNA随机引物使用聚合酶能读取尿嘧啶核苷酸合成含特定序列标签的DNA。然后用这些标签通过PCR在原始DNA链的5′和3′端添加接头序列。表观基因组库被稀释并加载到cBot DNA集群生成系统。聚类生成完成后,将2个样本转移到Illumina HiSeq 2000平台(北京诺禾致源公司)进行测序。所有步骤均按照制造商的说明进行。
1.5 BS-Seq数据分析
首先对Illumina测序获得的原始数据进行处理,过滤掉含有接头序列、未知或低质量碱基的reads, 然后用比对软件Bowtie 2 (http://bowtiebio.sourceforge.net/bowtie2/index.shtml)将唯一的净reads映射到水稻参考基因组(https://phytozome.jgi.doe.gov/pz/portal.html#info?alias=Org_Osativa)。基因组元素的鉴定采用Li等[12]方法进行。
利用BS-Seq数据检测单个碱基的甲基化状态、甲基化胞嘧啶位点的数量以及每个基因组原色的甲基化比例。甲基化水平(%)由每个甲基化胞嘧啶(mC)的读数除以胞嘧啶的总读数得到。差异甲基化区域(DMRs)包含至少5个CG (CHH或CHG)位点,甲基化水平发生2倍变化(Fisher精确检验,P≤0.05)。在多次试验中,采用错误发现率(FDR)方法确定P值的阈值。使用R包进行基础数据操作和统计分析。
当1个基因上至少有1个DMR时,该基因被认为是差异甲基化基因(DMG)。使用Blast2GO软件获取每个DMG的GO信息,利用WEGO网站(http://wego.genomics.org.cn/)上传每个DMG的注释信息,并根据功能对DMGs进行分类。功能意义的GO富集分析应用超几何测试将所有DMGs映射到GO数据库中,在与全基因组相比的差异甲基化基因中搜索显著富集的GO术语[13]。
与全基因组背景相比, KEGG通路富集分析可确定DMGs中显著富集的代谢通路或信号转导通路。计算公式和应用程序与GO分析相同。随后,以FDR (q值)≤0.05作为阈值来确定DMGs中最显著富集的通路。
使用MapMan软件(http://mapman.gabipb.org)描述与DMGs相关的生物途径[14]。
为提供高或低DMGs的描述,使用Cluster软件和JAVA TreeView软件进行聚类分析。将一份包含2个样本中DMGs及其甲基化比例的文件上传到Gene Cluster 3.0。经过数据过滤和日志转换后,采用平均连锁法对基因和阵列进行分层聚类。利用Cluster软件生成的格式文件结合JAVA TreeView软件,可直观地观察图形化显示的聚类结果。
1.6 RNA提取和转录组测序
使用TaKaRa RNAiso Plus (总RNA提取试剂,日本TaKaRa公司), 参照试剂盒的标准操作程序,从用于甲基组分析的同一绿苗和黄化苗中分离总RNA。使用NanoDrop ND-2000分光光度计(美国赛默飞世尔公司)和Agilent生物分析仪(2100, 美国安捷伦公司)检测RNA浓度和质量。使用RNA Clean XP Kit (Cat#A63987, 美国见克曼库尔特公司)和RNase-Free DNase Set (Cat#79254, 德国凯杰公司)纯化总RNA。
每个样品使用5 μg RNA进行文库构建。文库测序由上海伯豪生物公司完成。利用IlluminaHiSeq 2000/2500、MiSeq、Next Generation Sequencing (NGS)技术对绿苗和黄化苗的cDNA进行测序。移除接头序列和低质量reads, 执行端测序。使用HISAT和Bowtie2工具将净reads比对到水稻基因组注释项目(http://rice.plantbiology.msu.edu/)。基因表达分析使用RESM软件计算FPKM值。差异表达基因(DEGs)采用NOISeq软件进行筛选,筛选标准为fold change≥2且P≤0.05。利用GO和KEGG工具对DEGs进行分析。
1.7 RNA提取和RT-qPCR分析基因表达
用TaKaRa RNAiso Plus (总RNA提取试剂)从生长14 d的绿苗和黄化苗幼叶中提取和纯化总RNA。用TaKaRa PrimeScriptTM RT Reagent Kit合成第1链cDNA, 用不含RNase的ddH2O稀释反转录产物。RT-qPCR分析使用TaKaRa SYBR Premis Ex Taq II (Perfect Real Time)试剂盒和ABI 7300实时定量PCR系统(美国Applied Biosystems公司)。泛素蛋白UBQ引物的UBQ-1作为内参基因。采用循环阈值(Ct) 2-ΔΔCt方法计算每个样本中所选基因的相对表达量。进行3次独立的生物学重复。用于RT-qPCR的基因特异性引物见表1。
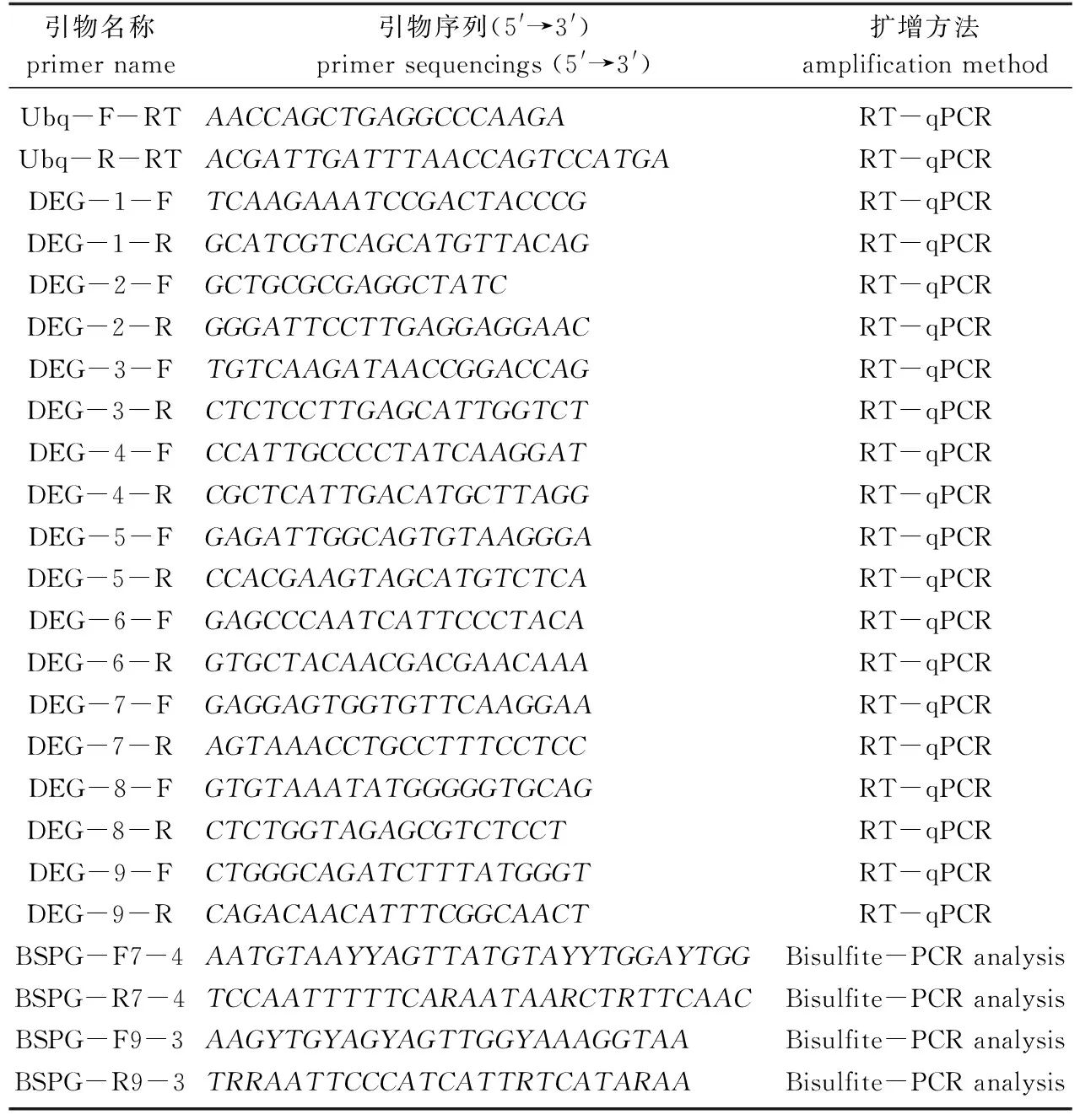
表1 RT-qPCR和重亚硫酸盐测序试验使用的引物Tab.1 Primers used in RT-qPCR experiments and Bisulfite-PCR analysis
1.8 DNA提取与重亚硫酸盐测序
从生长14 d的绿苗和黄化苗幼叶中提取750 ng的DNA, 抽提的DNA用ZYMO甲基化试剂盒进行亚硫酸氢盐处理。使用Free Bisulfite Primer Design Tool (https://www.zymoresearch.com/ pages/bisulfite-primer-seeker)设计所选基因序列的引物。简并引物使用Ex Taq Hot Start (日本TaKaRa公司)从处理过的DNA中扩增目标片段。扩增产物使用Axygen凝胶提取试剂盒(美国Axygen公司)纯化,所有片段连接到pMDTM18 T载体(日本TaKaRa公司)中,挑选至少6个独立的单克隆由尚亚(杭州)生物技术公司测序。然后使用Kismeth (http://katahdin.mssm.edu/Kismeth)进行甲基化分析[15]。所用引物见表1。
2 结果与分析
2.1 水稻花药培养中黄化苗的形成
采用日本晴(OryzasativaL. subspjaponicacv. Nipponbare)进行水稻花药培养。花药形成愈伤组织,愈伤组织再分化成芽和根,逐渐发育成幼苗。在这一过程中会形成大量的黄化苗(图1.A、B), 2周后,绿苗和黄化苗被转移到再生培养基中(图1.C)。102根试管内的1 126粒花药形成293个愈伤组织,最终形成269株幼苗,其中绿苗为177株,黄化苗为92株,黄化苗率为34.20%。

图1 绿苗和黄化苗的表型和叶绿体超微结构分析*Fig.1 Phenotype and Chloroplast ultrastructure of green and etiolated seedlings* A. 花药培养愈伤组织诱导; B. 愈伤组织再生形成绿苗和黄化苗; C. 绿苗和黄化苗; D. 绿苗叶绿体超微结构(标尺=2 μm); E. 绿苗叶绿体超微结构(标尺=0.5 μm); F. 黄化苗叶绿体超微结构(标尺=2 μm); G. 黄化苗叶绿体超微结构(标尺=0.3 μm); H. 绿苗和黄化苗SPAD叶绿素值(0~100无单位); I. 绿苗和黄化苗叶绿素含量为分光光度计的测定值。* A. induction of callus from cultured rice anthers; B. regenerated rice green and etiolated seedlings from callus; C. green and etiolated seedlings; D and E. ultrastructure of the chloroplast of green seedlings, bar=2 μm, bar=0.5 μm, respectively; F and G. ultrastructure of the chloroplast of etiolated seedlings, bar=2 μm, bar=0.5 μm, respectively; H. SPAD chlorophyll value (a unitless index from 0 to 100) in green and etiolated seedlings; I. chlorophyll content measured by spectrophotometer.
与绿苗相比,黄化苗的SPAD值显著降低,说明相对叶绿素含量较低(图1.H)。利用细胞学鉴定技术,选择二倍体的绿苗和黄化苗开展研究。植物光合色素主要分为叶绿素a、叶绿素b和胡萝卜素等,位于叶绿体的类囊体膜上。黄化苗叶片上的叶绿素a、叶绿素b和胡萝卜素含量显著降低(图1.I)。透射电镜观察发现,绿苗绿叶中叶绿体结构完整,可见类囊体结构和明显的片层(图1.D、E), 但在黄化苗的幼叶中,类囊体片层不明显,液泡占据细胞(图1.F、G)。推测黄化苗的形成可能与叶绿体合成相关基因表达受到抑制有关。
2.2 绿苗和黄化苗的全基因组甲基化分析
对来自水稻花药培养的绿苗和黄化苗样品进行单碱基亚硫酸氢盐测序,从每个样本中过滤出大约7 780万和6 070万个净reads。绿苗和黄化苗样品的净率均超过91%, 表明测序质量较高,数据可靠,可用于高质量的全基因组甲基化分析。
绿苗与黄化苗之间DNA甲基化水平的全基因组整体差异不显著。在绿苗基因组中检测到超过15亿个胞嘧啶位点,在黄化苗基因组中检测到超过18.4亿个胞嘧啶位点,其中CG位点的mCs (Methylated C’s, 甲基化的C总数目)频率最高, CHG和CHH位点的的mCs频率最低(图2)。通过比较,绿苗和黄化苗在CHH位点略有差异,黄化苗在CG和CHH位点的甲基化水平明显高于绿苗(表2), 表明CG、CHG和CHH序列背景下的DNA甲基化受黄化的影响并不均匀。与绿苗相比,黄化苗在mCG、mCHG和mCHH位点检测到的mCs数量增加(图3.A-C), 表明在黄化苗形成过程中出现大量新的甲基化。绿苗和黄化苗之间的mC位点的比例在不同序列背景下也不同。结果发现mCHG位点的数量较mCHH位点的数量变化更多,而mCHH位点的数量较mCG位点的数量变化更多(图3.A-C)。

图2 绿苗和黄化苗的整个基因组中C、CpG、CHG和CHH的平均甲基化水平Fig.2 The average methylation levels of C, CpG, CHG and CHH in the whole genome of green and etiolated seedlings

图3 绿苗和黄化苗的DNA甲基化水平*Fig.3 The DNA methylome of green and etiolated seedlings* A-C. 绿苗和黄化苗之间mCs在mCG、mCHG、mCHH位点的比较; D-F. 绿苗和黄化苗在每一背景下水稻基因组不同区域甲基化序列的百分比; R1. 启动子, R2. 5′-非翻译区, R3. 外显子, R4. 内含子, R5. 3′-非翻译区, R6. 基因间区, R7. 相关转座因子。* A-C. comparison of mCs between green and etiolated seedlings for mCG, mCHG, mCHH sites; D-F. total percentage of methylated sequences in different regions of the rice genome for each context in green and etiolated seedlings; R1. promoter, R2. 5′-UTR, R3. exon, R4. intron, R5. 3′-UTR, R6. intergenic, R7. TE-related.

表2 绿苗和黄化苗整个基因组中C、CpG、CHG和CHH的平均甲基化水平Tab.2 The average methylation levels of C, CpG, CHG and CHH in the whole genome of green and etiolated seedlings
基于检测到的甲基化位点,计算了3个序列背景(mCG、mCHG和mCHH)中绿苗和黄化苗的7个区域(启动子、5′-UTR、外显子、内含子、3′-UTR、基因间和TE相关区域)的平均甲基化水平(图3.D-F), 发现在黄化苗的基因间区中mCG位点的比例略高。有趣的是,黄化苗中mCHG位点启动子区、内含子区、3′-UTR和TE相关区域的相对比例分别高于绿苗,而黄化苗中mCHH位点启动子区、内含子区、3′-UTR 和基因间区相对比例均高于绿苗。总体而言,绿苗和黄化苗中mCG和mCHG的7个区域的水平高度一致,其中,基因间区甲基化比例最多,其次是外显子区域。为更好地理解绿苗和黄化苗的基因和转座因子中DNA甲基化的不同模式,将基因组划分为几个功能基因区域: 启动子区、转录起始位点上游2 kb区域(TSS)、基因体、转录终止位点(TTS)下游2 kb处(图4.A-D)。据分析,mCG的甲基化水平最高,mCHG次之,mCHH再次之。总体而言,绿苗和黄化苗的CG、CHG和CHH甲基化趋势无明显变化。

图4 基因特征和染色体的DNA甲基化模式*Fig.4 DNA methylation pattern in gene features and chromosomesanalysis* A、C分别表示绿苗和黄化苗标准化甲基化水平, 这些区域是由转录起始位点(TSS)上游的2 kb, 以及转录区和转录终止位点(TTS)下游的2 kb构成; B、D分别代表mCG、mCHG和mCHH背景下基因功能区域的DNA甲基化模式, a. 启动子, b. 5′-UTR, c. 外显子, d. 内含子, e. 3′-UTR; E、F分别代表绿苗和黄化苗12条染色体上mCG、mCHG和mCHH背景下基因表达水平、转录因子(TEs)密度与甲基化水平的关系。* A and C represent the normalized methylation levels of green and etiolated seedlings, respectively, the plots were generated from 2 kb upstream of the transcription start site (TSS) to 2 kb downstream of the transcription termination site (TTS); B and D represent the DNA methylation patterns of gene functional regions in mCG, mCHG and mCHH environments in green and etiolated seedlings, respectively, a. promoter, b. 5′-UTR, c. exon, d. intron, e. 3′-UTR; E and F represent the circular plots of gene expression levels, transposable element (TE) density and methylation levels in mCG, mCHG and mCHH environments in rice green and etiolated seedlings, respectively.
为进一步确定12条染色体的DNA甲基化水平(mCG、mCHG和mCHH序列背景)转座元件(TEs)和基因表达之间的相关性,分析了染色体位置关系、基因注释和TEs信息,结果表明,在染色体水平上绿苗和黄化苗的DNA甲基化水平并没有明显的差异(图4.E、F)。然而在不同序列背景下,mCG的甲基化水平最高,而mCHH的甲基化水平最低。基因转录水平和DNA甲基化水平在每个染色体上的总体分布呈现出相反的模式(图4.E、F)。在水稻基因组中, DNA甲基化可能与TE密度呈正相关,与基因表达呈负相关,但仍未被大规模鉴定无论是在基因组规模上还是在特定位点上, CHH甲基化的水平往往低于CG或CHG甲基化水平[16]。其中, 1号染色体的0~5 Mb区域甲基化水平显著变化, 4号染色体的17~19 Mb区域甲基化水平显著变化, 9号染色体9~12 Mb以及11号染色体的6~8和26~28 Mb区域类似且甲基化水平显著变化。此外,每条染色体上的红色线(mCG)水平明显出现整体水平的上调,而每条染色体上的绿色线(mCHH)未发生明显变化(图5、6)。

图5 绿苗和黄化苗12条染色体基因组的DNA甲基化模式*Fig.5 Dynamics of DNA methylation in the genome of 12 chromosomes between green seedlings and etiolated seedlings* 黄化苗与绿苗的每条染色体上甲基化水平差异的比值; 红色代表mCG, 蓝线代表mCHG, 绿线代表mCHH。* tthe ratio of differences in methylation levels on each chromosome of etiolated seedlings to that of green seedlings; red linerepresents mCG, blue linerepresents mCHG, and green linerepresents mCHH.

图6 绿苗和黄化苗的染色体上差异甲基化位点及区域的分布*Fig.6 Distribution of differential methylation on chromosomes among green and etiolated seedlings* A和C分别为绿苗和黄化苗每条染色体上不同甲基化位点的比例, 红色代表高甲基化位点, 蓝色代表低甲基化位点; B和D分别为绿苗和黄化苗中每条染色体上不同甲基化区域的比例, 红色代表高甲基化区域, 蓝色代表低甲基化区域。* A and B indicate the number of differentially methylated sites on each chromosome in green and etiolated seedlings, respectively, red color indicates hypermethylated sites and blue color indicates hypomethylated sites; B and D indicate the number of differentially methylated regions (DMRs) on each chromosome in green and etiolated seedlings, respectively, red color indicates hypermethylated regions and blue color indicates hypomethylated regions.
2.3 绿苗和黄化苗的差异甲基化区域和GO富集分析
通过对绿苗和黄化苗DNA甲基化序列的全基因组比较,本研究鉴定了1 058个DMRs。在功能区中,与基因间相关的DMRs占比最大(图7.A)。从具有基因组特征的631个高甲基化DMRs和427个低甲基化DMRs中,鉴定出4 156个DMRs相关基因(图7.B)。在绿苗和黄化苗的CG位点处检测到2 769个DMRs相关基因,在CHG位点检测到1 095个DMRs相关基因,在CHH位点检测到292个DMRs相关基因,共获得1 907个启动子相关的DMGs和2 249个基因本体相关的DMGs, 这与早前的研究[17]结果一致,甲基化差异主要集中在CG位点,表明基因本体的DNA甲基化在整个基因组中受到广泛调控,启动子区域的甲基化相对次之。

图7 绿苗和黄化苗甲基化差异区分布*Fig.7 Distribution of differentially methylated regions in green and etiolated seedlings* A. 基因组上高甲基化和低甲基化区域(DMRs)的分布; B. 绿苗和黄化苗的不同甲基化序列背景下相关基因的基因特征分布; C和D分别表示启动子和基因体相关基因的GO分析。* A. distribution of hyper-and hypomethylated regions (DMRS) on the genome; B. the distribution of different methylated context-associated genes among the gene features in etiolated/green seedlings; C and D represent the Gene ontology (GO) analysis of genes associated with promoters and gene body, respectively.
为描述基因及其产物的性质,使用WEGO对DMGs进行功能分类。GO分析显示,相对于绿苗,黄化苗参与多种分子功能和生物过程,包括有机物代谢过程、初级代谢过程、细胞代谢过程、催化活性、杂环化合物结合和细胞解剖实体(图7.C), 表明与基因本体相关的甲基化以及与启动子相关的甲基化各自占有一定比例。CHH序列背景的甲基化是基因本体内的主要甲基化位点,而CG序列背景甲基化是基因本体内的主要甲基化位点。通过对DMRs相关启动子的GO分析,发现与参与有机物质代谢过程、初级代谢过程、细胞代谢过程、细胞周期过程、结合、催化活性和杂环化合物结合等通路相关基因有关(图7.D)。
2.4 绿苗和黄化苗的差异基因表达分析
有研究[18-19]显示,基因表达与DNA甲基化水平呈负相关性。本研究基于RNA-Seq分析了绿苗和黄化苗之间的差异基因表达,测序数据表明测序结果良好、数据质量合格,可用于基因组比对。绿苗和黄化苗样品中唯一的比较reads分别为4 768万和6 731万。通过测序获得了2个样品的每百万个作图读数的外显子模型的每千碱基片段(FPKM)和倍数变化(FC)值。由于CG序列背景是基因本体内的主要甲基化位点,因此,差异基因表达分析以CG序列背景进行,发现绿苗和黄化苗中大多数基因在CG位点的表达水平相似,仅有411个基因上调, 646个基因下调(图8.A)。
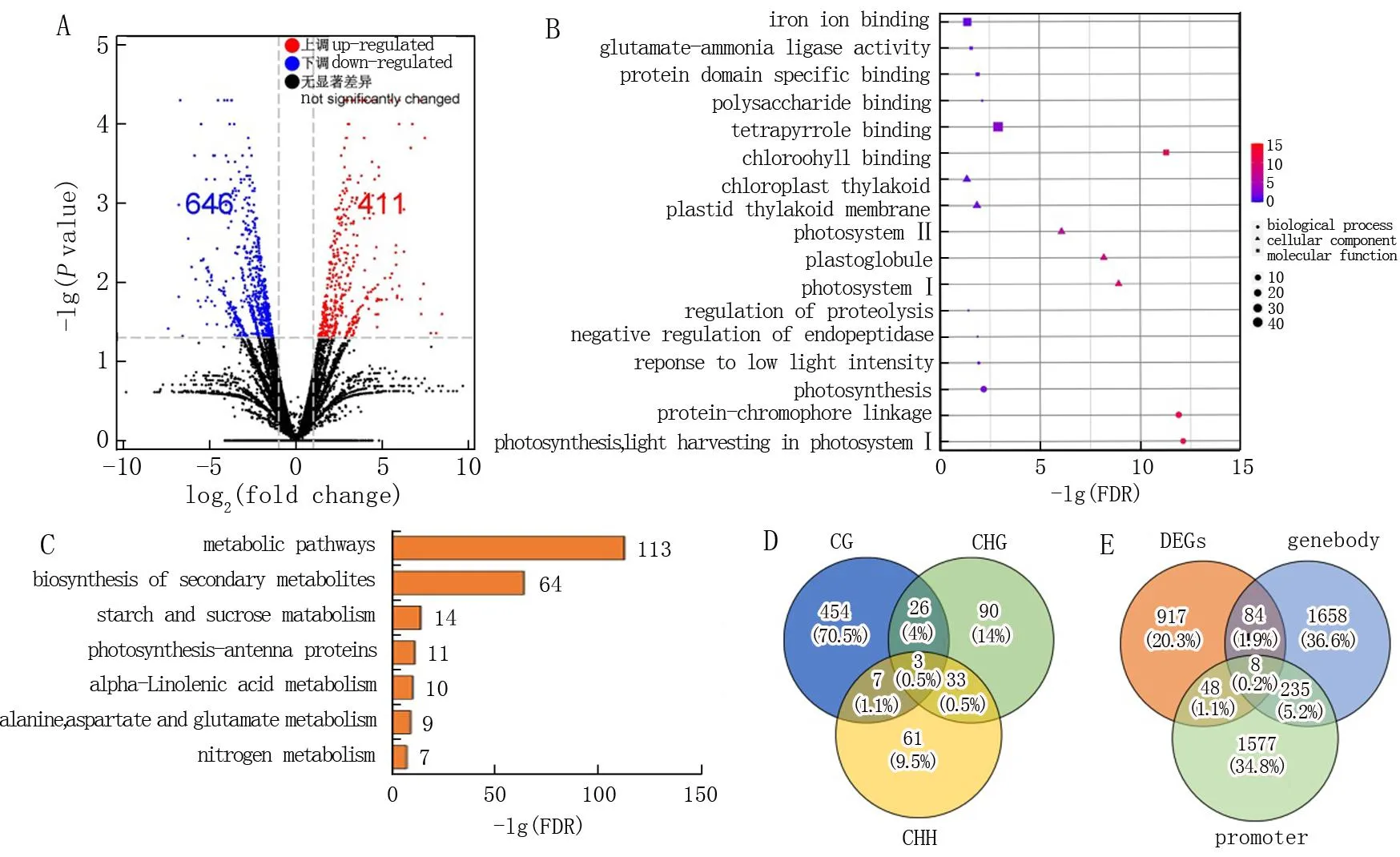
图8 绿苗和黄化苗基因表达差异及GO富集分析*Fig.8 Differential gene expression among green and etiolated seedlings and GO enrichment analysis* A. CG序列背景下绿苗和黄化苗之间基因转录的火山图, DEGs的阈值为P≤0.05且倍数>2, 红点和蓝点分别代表表达上调和下调的基因, 黑点代表无显著表达的基因; B. DEGs的GO富集分析; C. DEGs的KEGG通路富集分析; D. 不同序列背景下甲基化变化的DEGs比较; E. 在一种或多种序列背景下甲基化差异的基因与DEGs之间的比较。“Gene body”和“promoter”分别代表转录区和基因上游区域1 kb,“DEGs”代表绿苗和黄化苗RNA-Seq中的差异表达基因。* A. volcano plot indicating gene transcription between green and etiolated seedlings in CG context, DEGs were identified by using a threshold of P≤0.05 and fold change>2, red and blue dots represent genes with up-and down-regulated expression, respectively, whereas the black dots represent the genes with no significant expression; B. GO enrichment analysis of differentially expressed genes; C. KEGG pathway enrichment analysis of DEGs; D. comparison of DEGs according to methylation variations among sequence contexts; E. comparison of genes differentially methylated at one or more sequence contexts and differentially expressed between green and etiolated seedlings. ‘Gene body’ and ‘promoter’ represent the transcribed region and the 1 kb upstream region of genes, respectively; ‘DEGs’ represents genes that are differentially expressed genes in RNA-seq among green and etiolated seedlings.
利用GO分析绿苗和黄化苗在CG位点的1 057个DEGs, 结果表明,在生物过程方面主要参与多糖结合、蛋白水解调控、内肽酶活性负调控、对低光强度刺激的响应、光合作用、蛋白质-色素细胞联动、光采集在光系统I中的反应; 细胞组分方面主要与类囊体膜、质体类囊体膜、光系统Ⅱ、质体小球和光系统Ⅰ有关; 分子功能方面主要与铁离子结合、谷氨酰胺-氨连接酶活性、蛋白质结构域特异性结合、四吡咯结合和叶绿素结合有关(图8.B)。KEGG富集分析发现, DEGs在代谢途径显著富集,包括次生代谢物生物合成途径、淀粉和蔗糖代谢、光合作用-天线蛋白、α-亚麻酸代谢、丙氨酸-天冬氨酸-谷氨酸代谢和氮代谢(图8.C) GO与KEGG富集分析表明与光合作用相关的途径均与蛋白质表达的改变有关。
试验鉴定出的644个基因在绿苗和黄化苗之间的甲基化和基因表达均表现出显著差异(以下称为共差异基因), 只有0.5% (644个基因中的3个)在所有3个序列背景中存在差异甲基化(图8.D), 分别是编码叶绿体酶基因LOC_Os06g01850(OsLFNR1, 铁氧还蛋白-NADP+氧化还原酶)、基因LOC_Os01g41710(假定表达基因叶绿素A-B结合蛋白)和基因LOC_Os02g10390(假定表达基因叶绿素A-B 结合蛋白), 其中OsLFNR1在光合作用电子传递链的最后步骤中发挥功能[20]。这表明甲基化对叶绿素合成过程中的转录水平具有重要影响。此外,绿苗和黄化苗差异表达的644个基因中, 490个基因含有mCG位点, 122个基因含有mCHG位点, 74个基因含有mCHH位点。进一步比较显示,在绿苗和黄化苗中,只有8个基因同时在基因本体和启动子区域发生不同程度的甲基化(图8.E), 表明基因本体和启动子区域的甲基化在很大程度上是由独立的机制分别调节的。
2.5 绿苗和黄化苗光合作用途径的共差异基因
叶绿体生物合成在光合作用和植物生长中发挥着至关重要的作用,以响应环境变化。为研究绿苗和黄化苗之间的差异基因表达,重点研究参与叶绿体生物合成相关途径的共差异基因。根据GO和KEGG富集分析,确定了9个与绿苗和黄化苗光合作用途径有潜在关系的基因,这些基因是甲基化上调而表达下调的基因(表3), 其中包括编码叶绿素生物合成酶的基因OsPORA[21], 在水稻光合作用和叶绿体发育中发挥功能的基因OsTLP27[22], 编码在光合作用电子传递链的最后步骤中发挥功能的一个重要的叶绿体酶基因OsLFNR1[22]参与光系统而影响光合作用能力的基因OsNPH1a[23], 通过影响水稻叶片光合率和糖代谢而参与叶绿素积累、叶绿体发育以及植株生长的基因OsAld-Y51[24], 具有转氨酶作用的类Ⅰ和类Ⅱ结构域含蛋白质的表达基因LOC_Os07g01760, 以及假定的类囊体管腔 29.8 ku 蛋白质的编码基因LOC_Os12g08830和参与光系统Ⅱ组装的基因LOC_Os07g05360。

表3 9个可能与光合作用相关的基因Tab.3 Nine genes potentially related to photosynthesis pathways
通过RT-qPCR分析发现,上述9个DMGs中有8个基因的表达水平下降,这与转录组测序结果一致(图9.A)。为验证甲基化模式的结果,随机选择2个基因使用重亚硫酸盐测序分析。基于BS-Seq结果,重点研究参与光合过程光系统II组装的基因LOC_Os07g05360和参与结构域含蛋白的表达基因LOC_Os07g01760, 结果显示这2个基因的甲基化水平分别为18.145%和91.891%, 均处于高甲基化水平,重亚硫酸盐测序的结果与甲基化组分析的结果一致(图9.B), 表明测序结果可靠。

图9 9个DMGs的RT-qPCR和重亚硫酸盐测序验证*Fig.9 Expression level of nine DMGs validated by RT-qPCR and bisulfite-PCR* A. 9个DMGs在绿苗和黄化苗的RT-qPCR分析,所有的RT-qPCR反应, 3个生物重复; B. 绿苗和黄化苗LOC_Os07g05360和LOC_Os07g01760的DNA甲基化水平。* A. RT-qPCR analysis of nine DMGs in green and etiolated seedlings, all RT-qPCR reactions were performed with three biological replicates; B. DNA methylation levels of LOC_Os07g05360 and LOC_Os07g01760 in green and etiolated seedlings.
3 讨论
表观遗传修饰在植物逆境响应机制中起着重要作用。环境刺激如盐和渗透胁迫可导致某些基因编码区的去甲基化,从而激活其表达。在某些不利条件下,非生物胁迫也会导致DNA甲基化和基因表达模式的改变。有研究[25]表明,植物响应高温、低温、干旱、盐碱、重金属等非生物胁迫过程中,通过全基因组或特定基因DNA甲基化水平变化,进而调控抗逆基因的表达以更好地适应生存环境。此外,在转基因过程中, DNA甲基化参与调节植物开花、花器官形成和植物基因沉默[26-27]。水稻花药培养简单、省时、适应性广泛,但高频的黄化现象严重影响着水稻的育种进程。叶绿体生物合成是一个非常复杂的过程[28], 通过一系列协同反应,且有多种酶催化完成的[29]。叶绿体负责光合作用以及激素与代谢物的产生[30-31], 这些激素和代谢物是由存在于植物分生组织未成熟细胞中前质体发展而来的。叶绿体的生物发生涉及质体和核编码基因[32]的协调功能。叶绿体的生物发生过程中伴随着叶绿素、叶绿体蛋白、类胡萝卜素和酯类的生物合成,以及光合蛋白复合体的组装,包括捕光复合体(LHC)、光系统Ⅰ (PSⅠ)、光系统Ⅱ (PSⅡ)、细胞色素f/be (cytf/bs)和三磷酸腺苷(ATP)合酶[30,33]。水稻花药培养的绿苗和黄化苗,经转录组分析,发现通过RT-qPCR试验鉴定出9个与光合途径相关的表达下调的基因,同时经重亚硫酸盐测序分析,随机鉴定其中2个DNA甲基化上调的基因。其中,LOC_Os07g053600[34]是光反应和卡尔文循环中起作用的基因。LOC_Os07g01760[35]是一种氨基酸转移酶,推测在多胺降解中起作用,这些基因在光合途径中均起着至关重要的作用。
通过对水稻花药培养获得的绿苗和黄化苗观察和细胞超微结构分析发现,黄化苗叶片黄化,叶绿体发育不完整,叶绿素含量极少,生长发育受阻等,造成水稻幼苗不能进行正常的光合作用,无法正常生长。同时,经验证的9个与光合作用相关的基因表达下调,故推测DNA甲基化会影响叶绿体生物合成相关基因,进而引起叶绿体生物合成受阻,导致高频黄化苗的产生。本研究结果说明, DNA甲基化与基因表达之间的关系比早前认识要复杂,而启动子的甲基化通常与基因表达抑制有关,有关基因甲基化的作用则更不确定,既有正向也有反向的调控关系。同时,本研究对水稻绿苗和黄化苗叶片差异表达基因和差异甲基化基因的功能注释和富集分析有助于探讨DNA甲基化在水稻叶片中各代谢通路的表达与调控,为进一步研究DNA甲基化对花药培养中黄化苗的产生以及光合作用的影响机制提供理论基础。
