第一代TRK抑制剂拉罗替尼关键手性胺中间体(R)-2-(2,5-二氟苯基)吡咯烷的合成工艺研究
2024-01-10裴超陈玉琴吴筱斐柴文静杨爱青王磊磊刘团伟魏金建孙洪宜张志德
裴超,陈玉琴,吴筱斐,柴文静,杨爱青,王磊磊,刘团伟,魏金建,*,孙洪宜*,张志德*
(1.中瀚(齐河县)生物医药科技有限公司,山东 德州 251114;2.山东师范大学化学化工与材料科学学院,山东 济南 250014;3.齐鲁工业大学(山东省科学院),山东省科学院生态研究所,山东省应用微生物重点实验室,山东 济南 250014)
Tropomycin-related kinase (TRK)为神经营养因子酪氨酸激酶受体,隶属于受体酪氨酸激酶家族[1-3]。大量的研究表明TRK信号转导通路的活化与肿瘤的发生、发展有很强的相关性,在神经细胞瘤、前列腺癌、乳腺癌等中均发现了活化的TRK信号蛋白,近几年来多种TRK融合蛋白的发现更显示了其促进肿瘤发生的生物学功能[4-5]。如果能抑制激酶活性,就能抑制癌症生长,TRK抑制剂的原理就是抑制激酶的活性[6]。
美国Loxo Oncology公司开发的第一代TRK抑制剂拉罗替尼(图1a)可有效治疗17种肿瘤,针对多种基因融合的患者有效率高达76%,包括12%的患者肿瘤完全消失,该药于2018年被FDA批准上市,2022年4月13日获得中国国家药品监督管理局批准正式上市[5-6]。拉罗替尼具有口服、针对17种不同肿瘤均可使用的广谱靶向抗癌药,临床实验结果证明拉罗替尼对不限年龄的TRK融合癌症患者具有持久的抗肿瘤作用、良好的耐受性和很小的副作用等优势,是TRK基因突变癌症患者的第一选择[6]。然而拉罗替尼的价格非常昂贵,成人服用拉罗替尼胶囊30 d的费用约为32 800美元(约合人民币20万元),儿童服用其口服液的费用每月至少11 000美元。患者肿瘤部位的表面积越大,使用剂量越大,费用越高,一般患者根本望尘莫及,而科技的进步无法惠及万千患者的根本在于原料药的产业化工艺。拉罗替尼工业化的一个难点在于其关键手性胺片段(R)-2-(2,5-二氟苯基)吡咯烷(图1b)的低成本制备,目前已报道的该中间体合成路线均通过手性诱导试剂或格氏试剂参与的反应合成[7-10],通用的合成工艺路线如图2所示。
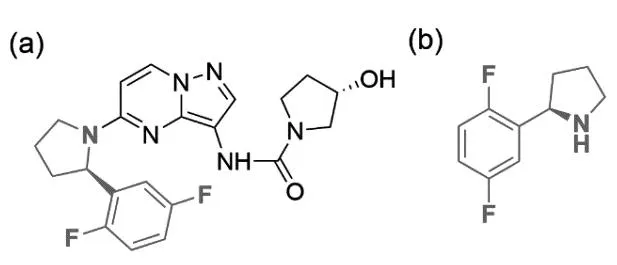
图1 拉罗替尼和其关键手性片段(R)-2-(2,5-二氟苯基)吡咯烷的化学结构式
路线1:

图2 已报道的(R)-2-(2,5-二氟苯基)吡咯烷的合成路线
路线1和2均采用手性诱导试剂(S)-2-叔丁基磺酰胺合成手性胺中间体,该试剂昂贵且具有难闻的臭味,不符合现有的环保要求,难于规模化应用;此外,路线1使用昂贵的三乙基硼氢化锂(LiBEt3)还原脱除叔丁基亚磺酰亚胺,需在-78 ℃下进行,总收率仅为36%,生产成本极高,难以实现产业化[7];路线2使用昂贵的2,5-二氟苯甲醛及2-(2-溴乙基)-1,3-二氧杂环己烷,且需要危险的格氏反应工艺,难于实现安全化,该路线原材料生产成本极高,难以规模化生产[8];路线3采用昂贵的手性试剂诱导手性中间体的合成,且用到硼烷二甲硫醚,具有难闻的臭味,不符合现有的环保要求,该路线也使用危险的格氏反应工艺,难以实现操作安全化和规模化应用[9-10]。以上报道的三条工艺路线虽然使用手性诱导催化剂,但所得的光学纯度(ee值)较低,仍需对手性胺进行化学拆分才能达到≥98% ee值的质量要求,生产成本极高。
为解决以上报道合成路线中存在的共性问题,本论文研究了拉罗替尼关键手性胺中间体(R)-2-(2,5-二氟苯基)吡咯烷的创新合成路线(图3),该路线以廉价的2,5-二氟甲苯为起始原料,经氧化得到中间体1,经酯化生成中间体2,后经亲核取代、酸解、脱羧和环合一步法得到中间体3,后经还原得到消旋化物中间体4,再经化学拆分得到手性胺目标物,总收率为23%。该路线避免使用昂贵的原材料和试剂如手性诱导试剂、低温和无水无氧环境,更适合拉罗替尼关键手性胺中间体(R)-2-(2,5-二氟苯基)吡咯烷的产业化。
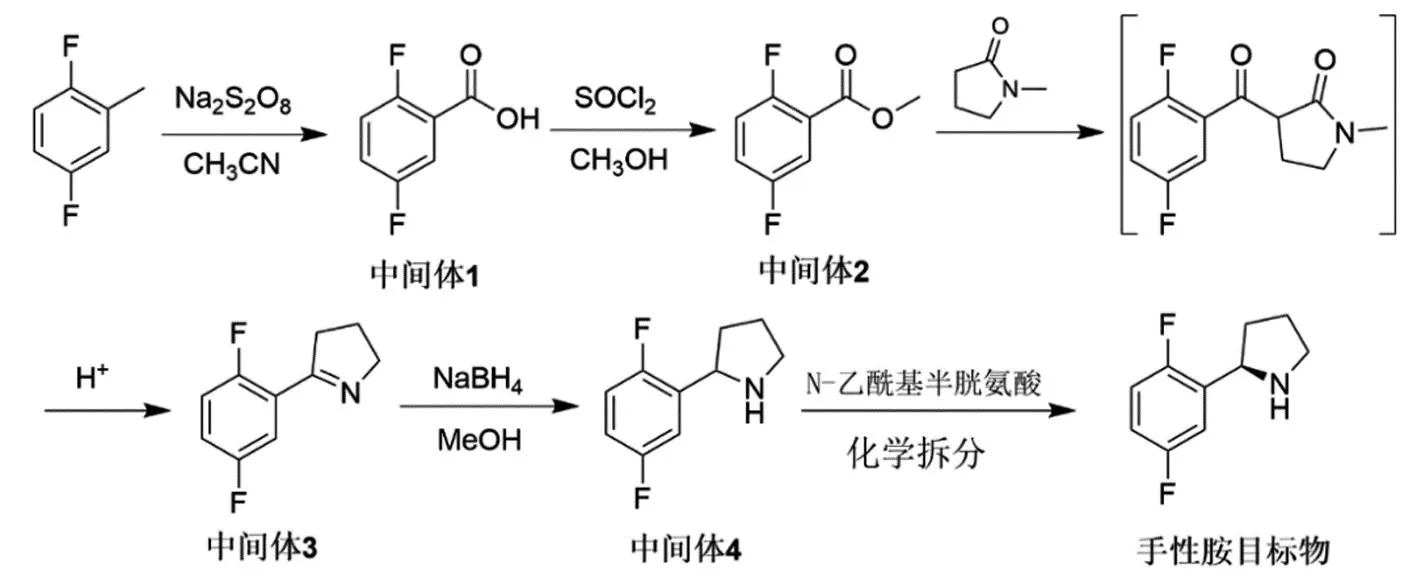
图3 本论文报道的(R)-2-(2,5-二氟苯基)吡咯烷的新合成路线
1 实验部分
1.1 仪器和试剂
400 MHz核磁共振谱仪(Bruker,Bruker Advance Ⅱ),高效液相色谱(Waters e2695)。
2,5-二氟甲苯、过硫酸钠、乙腈、氯化亚砜、甲醇、N-甲基吡咯酮、盐酸、硼氢化钠、N-乙酰基-D-半胱氨酸均为分析纯试剂,来源于国药集团。
1.2 化合物的合成
1.2.1 2,5-二氟苯甲酸(中间体1)的合成
将2,5-二氟甲苯(20 g,0.16 mol)溶于100 mL乙腈中,室温下缓慢加入过硫酸钠(45.7 g,0.19 mol),缓慢升温至60 ℃,TLC监测至反应完全(展开剂:正己烷),缓慢滴加水,析出淡黄色固体,抽滤、干燥得到23.5 g中间体1,收率95%,mp 132~134.1H NMR (400 MHz,d6-DMSO),δ: 13.54 (brs,1H,CO2H),7.60~7.63 (m,1H,Ar-H),7.51~7.55 (m,1H,Ar-H),7.37~7.42 (m,1H,Ar-H).19F NMR (372 MHz,d6-DMSO),δ: -116.07~-116.18 (m,1F),-118.04~-118.15 (m,1F)。
1.2.2 2,5-二氟苯甲酸甲酯(中间体2)的合成
将中间体1(22 g,0.14 mol)加入至110 mL甲醇中,室温搅拌下缓慢滴加氯化亚砜(20.2 g,0.17 mol),滴加完毕后,升温至70 ℃反应5 h,TLC检测反应结束(展开剂:乙酸乙酯/甲醇,体积比100∶1),减压蒸出溶剂,搅拌降温至0~5 ℃,滴加饱和NaHCO3(aq)调节pH值=7~8,加入二氯甲烷萃取(100 mL×2),有机相干燥,减压蒸干得到23 g中间体2,收率:95.8%。1H NMR (400 MHz,CDCl3),δ: 7.60~7.65 (m,1H,Ar-H),7.19~7.25 (m,1H,Ar-H),7.09~7.15 (m,1H,Ar-H),3.94 (m,3H,OCH3).19F NMR (372 MHz,d6-DMSO),δ: -115.42~-115.32 (m,1F),-118.12~-118.01 (m,1F)。
1.2.3 5-(2,5-二氟苯基)-3,4-二氢-2H-吡咯(中间体3)的合成
将N-甲基吡咯烷酮(13.8 g,0.14 mol)溶于100 mL乙腈中,降温至-5~0 ℃,缓慢加入叔丁醇钾(14.6 g,0.13 mol),搅拌10~15 min,将中间体2(20 g,0.12 mol)缓慢加入至上述反应液中,滴加完毕后,室温反应,TLC监测中间体2消失(展开剂:乙酸乙酯/正己烷,体积比1∶5),降温至室温后,向上述反应液中缓慢滴加15 mL浓盐酸,升温至70 ℃后保温反应6 h,调碱,减压去除溶剂,二氯甲烷萃取、干燥,减压除去溶剂得到浅黄色液体13.7 g,收率65.2%。1H NMR(400 MHz,CDCl3),δ: 7.22~7.27 (m,1H,Ar-H),6.91~6.97 (m,1H,Ar-H),6.83~6.87(m,1H,Ar-H),4.40(t,3H,J=7.6,CH2),3.13~3.22(m,1H),3.02~3.08 (m,1H),2.21~2.30 (m,1H),1.80~1.93 (m,1H),1.57~1.66 (m,1H).19F NMR (372 MHz,d6-DMSO),δ: -119.08~-118.98(m,1F),-124.87~-124.81 (m,1F)。
1.2.4 2-(2,5-二氟苯基)四氢吡咯(中间体4)的合成
将中间体3(13 g,0.07 mol)的甲醇溶液(130 mL)降温至0~5 ℃,分批加入NaBH4(3.4 g,0.09 mol)后室温反应,TLC监控反应,反应结束后,向体系中滴加水破坏NaBH4,减压蒸出溶剂,二氯甲烷萃取,干燥有机相,蒸干得到12 g中间体4,收率92%。1H NMR(400 MHz,CDCl3),δ: 7.63~7.68(m,1H,Ar-H),7.02~7.10(m,2H,Ar-H),4.00~4.03(m,2H,CH2),2.70~3.02(m,2H,CH2),2.00~2.08(m,2H,CH2).19F NMR(372 MHz,d6-DMSO),δ: -118.89~-118.71 (m,2F)。
1.2.5 (R)-2-(2,5-二氟苯基)四氢吡咯(手性胺目标物)的合成
将中间体4(10 g,0.05 mol)加入到50 mL异丙醇中,加入N-乙酰基-D-半胱氨酸(8.1 g,0.05 mol),升温80 ℃搅拌反应5 h,降温析晶,抽滤得到白色固体,将固体加入到水中,用2 mol/L NaOH(aq)调节碱性,二氯甲烷萃取,有机相干燥蒸干,重复上述操作一次,得到4.2 g浅黄色油状手性胺目标物,收率42%,经衍生方法HPLC检测得ee值为99.5%。
1.3 (R)-2-(2,5-二氟苯基)吡咯烷的液相检测方法
1.3.1 色谱条件
色谱柱:Xtimate®C18,4.6 mm×250 mm,5 μm P/N:00101-21043。
以0.01 mol/L乙酸铵/乙腈(体积比40∶60)为流动相,检测波长为254 nm;流速为1.0 mL/min;柱温为30 ℃;进样量为10 μL。
1.3.2 溶液配制
1.3.2.1 制备溶液
向1.0 g(R)-2-(2,5-二氟苯基)吡咯烷中加入30%氢氧化钠溶液,样品溶液pH值≥14,加入甲叔醚剧烈摇匀,静置分层,将上层溶液转移至干净的旋蒸瓶,加压浓缩除去甲叔醚,作为制备溶液。
1.3.2.2 供试品溶液
取制备溶液10 mg,加入20 mg衍生试剂N-α-(2,4-硝基-5-氟苯基)-L-丙氨酸,加20 mL乙醇溶解,90 ℃下加热回流2 min,浓缩除去乙醇,加入10 mL乙腈溶解,作为供试品溶液。
1.3.2.3 空白溶液
精密称定20 mg衍生试剂N-α-(2,4-硝基-5-氟苯基)-L-丙氨酸,加20 mL乙醇溶解,90 ℃下加热回流2 min,浓缩除去乙醇,加入10 mL乙腈溶解,作为空白溶液。
1.3.3 样品测试
取供试品溶液、空白溶液,分别注入液相色谱仪,记录色谱图。按公司内控标准,面积归一化计,ee值应不低于99.5%。
2 结果与讨论
2.1 化合物的合成
与前期报道的(R)-2-(2,5-二氟苯基)吡咯烷工艺路线相比,本文报道的新路线避免了使用危险的格氏反应、昂贵的手性诱导试剂、低温和无水无氧操作。以廉价的起始原料2,5-二氟甲苯经氧化得到中间体1,采用无机盐过硫酸钠作为氧化剂,避免使用了重金属氧化剂,简化了后处理步骤。第二步甲酯化采用传统的SOCl2/MeOH方法,为除去少量的中间体1,反应后处理需用弱碱如饱和NaHCO3溶液调剂pH值至7~8,pH值过高会导致中间体2的水解。本工艺路线的关键在于中间3的合成,借鉴最新报道的尼古丁合成路线,采用亲核取代、酸解、脱羧和环合反应一步法生成中间体3。N-甲基吡咯酮在强碱叔丁醇钾的存在下,形成碳负离子,进攻中间体2羰基,形成1,3-二羰基化物,该中间态无需进行分离,加酸导致吡咯酮开环,加热条件下脱羧,而在酸性条件下,羰基和胺基发生分子内环合生成中间体3。中间体4的合成采用经典的NaBH4还原即可高效实现,消旋体中间体4在N-乙酰基-D-半胱氨酸存在下经化学拆分即可高效得到单一光学手性的(R)-2-(2,5-二氟苯基)吡咯烷。
2.2 核磁共振氢谱分析
中间体1的1H NMR显示7.0~8.0为苯环上三个氢的特征峰,13.5为羧基氢特征峰,说明中间体1被成功合成;而中间体2的1H NMR 显示除7.0~7.5为苯环上的三个氢的特征峰外,3.8为甲酯甲基氢特征峰,中间体11H NMR中对应的13.5处羧基氢消失,说明甲酯化反应成功;中间体3的1H NMR在2.0~4.0范围内为吡咯环的三组亚甲基氢特征峰,与中间体2的1H NMR进行对照,环合反应发生且形成了中间体3;中间体4的1H NMR 在7.0~7.5为苯环上的三个氢的特征峰,4.2为与苯环相连的苄位氢特征峰,2.7为氨基氢特征峰,其他为吡咯环的三组亚甲基氢特征峰,说明还原反应顺利进行,且生成了中间体4,目标R型手性胺的1H NMR与中间体4相同。
2.3 纯度分析
经本文工艺路线合成的(R)-2-(2,5-二氟苯基)吡咯烷的HPLC谱图如图4所示,6.8 min处对应目标R构型,纯度为99.82%,7.7 min为杂质S异构体,纯度为0.18%,总体ee值为99.64%,远大于现有技术中的98%ee值的质量标准,完全符合现有的原料药申报质量规范对异构体限度的控制要求。
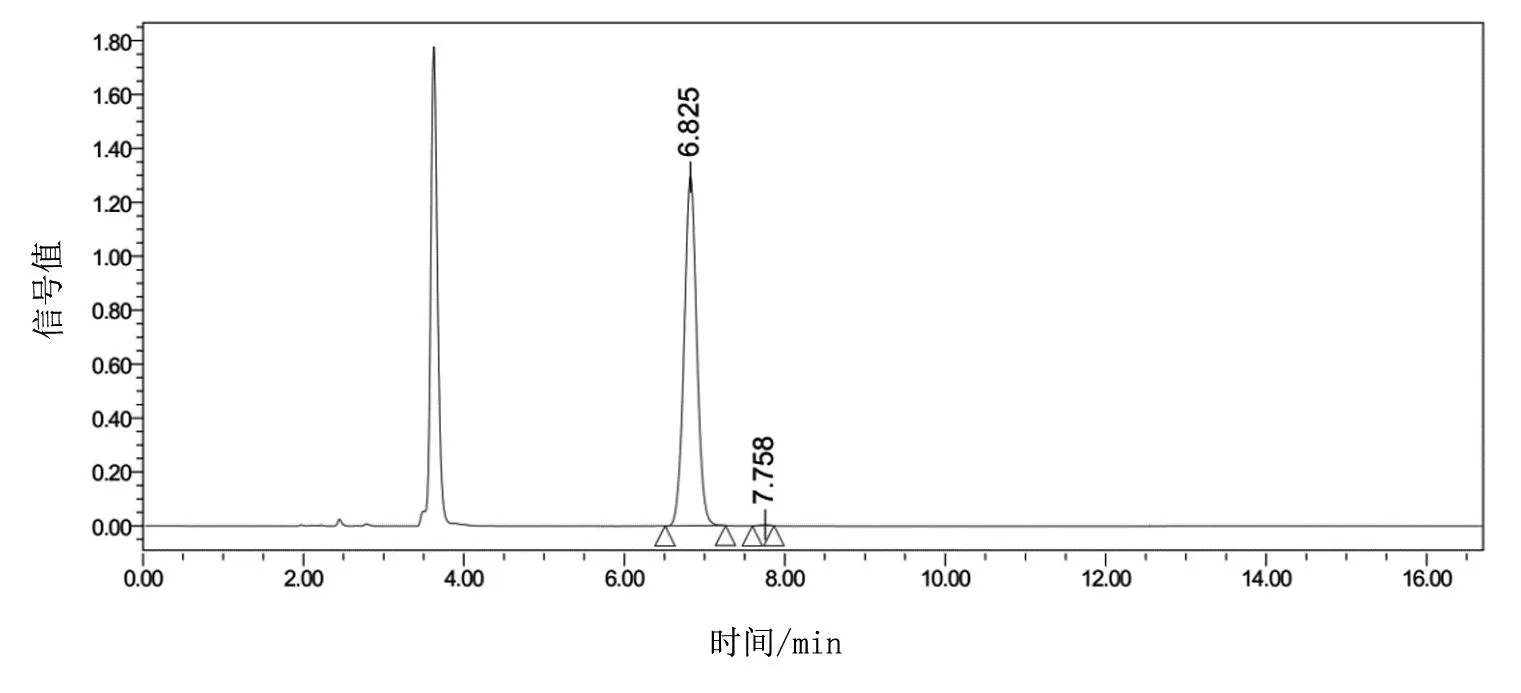
图4 (R)-2-(2,5-二氟苯基)吡咯烷的HPLC检测结果,6.825 min处对应的
化合物为R型异构体的纯度,7.758min处对应的杂质为S型异构体
3 结论
报道了拉罗替尼关键手性片段(R)-2-(2,5-二氟苯基)吡咯烷的新合成工艺路线,即以廉价的2,5-二氟甲苯为起始原料,经氧化、甲酯化、后经亲核取代、酸解、脱羧和环合一步法得到亚胺环合物,经还原得到消旋化物,再经化学拆分得到手性胺目标物,总收率为23%,所得(R)-2-(2,5-二氟苯基)吡咯烷的光学纯度为99.64%,远大于现有技术中的98%ee值的质量标准,完全符合现有的原料药申报质量规范对异构体限度的控制要求。该路线避免了使用危险的格氏反应、昂贵的手性诱导试剂、低温和无水无氧操作,更适合拉罗替尼关键。
