分子拥挤策略下硝苯地平表面印迹材料的制备与性能研究
2024-01-10赖申枝谢俊英
赖申枝,谢俊英
(湖南化工职业技术学院 制药与生物工程学院,湖南 株洲 412000)
二氢吡啶类钙离子通道抑制剂是一类降压作用好、临床使用效果好、上市品种丰富的心脑血管性疾病的治疗药[1]。硝苯地平是第一代二氢吡啶类钙离子拮抗剂[2],能干扰心血管平滑肌的兴奋收缩从而起到扩张血管作用,对不同类型的高血压均表现出较好的疗效,在高血压、心绞痛、心律失常的治疗方面得到大众认可[3]。近年来,某些不法商贩通过在保健品或食品中添加硝苯地平等违禁药物以达到降血压的效果,从而引发系列不良反应并耽误疾病的治疗。
目前对硝苯地平的分离检测方法主要有薄层色谱法[4]、高效液相色谱法[5-6]、紫外可见分光光度法[7]、液相色谱-质谱联用法[8]和毛细管电泳法[9]等。分子印迹技术[10]是以目标分子作为模板,制备与目标分子在三维结构、官能团、电荷等方面匹配性高的聚合物,因此制备的聚合物具有对目标分子的特异性识别和选择性吸附能力,且其性能稳定,重复使用性能较好[11],在复杂样品分离检测方面被广泛应用。分子拥挤试剂可以促进分子印迹反应过程形成稳定的三维结构,提高模板分子与功能单体之间的相互作用[12]。基于表面分子印迹技术[13]所制得的分子印迹聚合材料其识别位点存在于材料表面,能有效克服传统印迹法中模板分子包埋过深或过紧导致印迹效率不高的缺点,适用于复杂样品中特定成分的快速吸附分离[14]。
以硝苯地平为模板,基于表面分子印迹技术和分子拥挤技术,制备了一种硝苯地平表面分子印迹材料。以扫描电镜、透射电镜对材料进行形貌表征,并通过傅里叶变换红外光谱法等验证表面分子印迹层的成功涂覆。对聚合物的饱和吸附量、吸附动力学、吸附选择性和重复使用性能进行了考察。制备的表面印迹材料具有对竞争底物较好的选择性,在复杂样品组分的分离检测中有一定的应用前景。
1 实验材料和方法
1.1 仪器与试剂
Nicolet6700型红外光谱仪(日本Shimadzu公司);JSM-6610LV型扫描电子显微镜(日本JEOL公司);Lambda 25型紫外-可见分光光度计(日本Hitachi公司);JEM-2100F型透射电子显微镜(日本JEOL公司);SHA-C型恒温震荡仪(江苏金坛市荣华仪器制造有限公司)。
硝苯地平、苯磺酸氨氯地平、尼群地平、盐酸普萘洛尔、硅酸四乙酯(TEOS)、乙二醇二甲基丙烯酸酯(EGDMA)、聚苯乙烯购于上海阿拉丁生化科技股份有限公司,偶氮二异丁腈(AIBN)、甲基丙烯酸(MAA)购于国药集团化学试剂有限公司,十六烷基三甲基溴化铵(CTAB)购于安耐吉化学,其他试剂均为分析纯,实验用水为二次蒸馏水。
1.2 分子印迹聚合物制备
1.2.1 介孔二氧化硅微球的制备
向500 mL三口瓶中加入0.5 g CTAB,再加入50 mL无水乙醇与150 mL去离子水,搅拌下使其溶解,然后向上述混合液中添加8.0 mL氨水。反应条件为40 ℃恒温水浴且维持匀速机械搅拌,30 min后用恒压滴液漏斗缓慢滴加5.0 mL的TEOS,维持反应条件8 h以确保反应完全。然后将反应产物抽滤,用水/乙醇、乙醇分别各洗涤三次,产物抽滤后于真空干燥箱中50 ℃干燥24 h,即得介孔二氧化硅微球(mSiO2)。
1.2.2 表面分子印迹材料的制备
基于表面分子印迹技术,以介孔二氧化硅材料为载体,加入分子拥挤试剂,制备性能改进的表面分子印迹材料。将1 mmol硝苯地平溶于80 mL乙醇/氯仿(体积比1∶1)中并转入圆底烧瓶中,加入4 mmol 单体MAA,随后加入20 mg/mL的聚苯乙烯四氢呋喃溶液20 mL。往密闭的体系中通入氮气20 min,持续搅拌下,混合液于室温过夜,形成模板-单体-分子拥挤物的预聚物。将1.0 g 1.2.1制备的mSiO2材料加入到上述预聚液中,加入10 mmol交联剂EGDMA,在搅拌下通氮气30 min除氧。随后加入引发剂AIBN适量,并在持续搅拌下于60 ℃反应12 h。反应结束后,将反应产物抽滤,用甲醇/乙酸(体积比9∶1)洗脱液洗涤多次,用紫外-可见分光光度计检测洗脱液,直到检测不出硝苯地平。并用甲醇清洗去除残余的乙酸,抽滤。将沉淀于真空干燥箱中50 ℃干燥24 h,即得到表面分子印迹微球(MIP@mSiO2)。
制备非印迹微球(NIP@mSiO2)方法同表面分子印迹微球,制备过程中不加入模板分子硝苯地平。
1.3 表面分子印迹材料的吸附性能测试
1.3.1 静态吸附能力测定
用二氯甲烷配制含硝苯地平质量浓度为5,10,25,50,75,100,125,150 mg/L的系列待吸附液,称取20 mg表面分子印迹材料多份,分别加入到多个100 mL具塞锥形瓶,量取系列浓度的待吸附溶液40 mL分别加入各锥形瓶中,充分摇匀后置于恒温水浴振荡器中于25 ℃吸附60 min后抽滤。上清液经0.45 μm有机系微孔滤膜过滤,通过紫外-可见分光光度计测定(235 nm)硝苯地平的浓度。表面分子印迹材料的静态吸附量Q以吸附前后吸附液中硝苯地平浓度变化计算,见公式(1):
(1)
式中,Q为静态吸附量(mg/g),V为吸附测试液体积(L),C0为吸附前母液中硝苯地平的浓度(mg/L),Cx为吸附后吸附测试液中硝苯地平的浓度(mg/L),m为表面分子印迹材料的用量(g)。
根据印迹材料与非印迹材料静态吸附量的比值计算印迹因子IF,见公式(2):
(2)
1.3.2 吸附动力学实验
称取30 mg表面分子印迹材料10份,分别加入含硝苯地平质量浓度为75 mg/L的二氯甲烷溶液30 mL,充分振摇,置于恒温水浴振荡器中于25 ℃进行吸附,于不同的吸附时间(1,3,5,10,15,20,30,45,60,90 min)对上清液进行取样,按照1.3.1方法测定上清液中硝苯地平的浓度,根据上清液中硝苯地平浓度变化计算材料的吸附量随时间的变化情况。
1.3.3 选择性吸附实验
依照静态吸附法,测定MIP@mSiO2和NIP@mSiO2对硝苯地平及其结构类似物尼群地平、苯磺酸氨氯地平和竞争底物盐酸普萘洛尔的吸附量。分别配制质量浓度均为100 mg/L的硝苯地平、尼群地平、苯磺酸氨氯地平、盐酸普萘洛尔二氯甲烷/甲醇溶液(体积比1∶1),取40 mL置于100 mL具塞锥形瓶中,各自加入20 mg MIP@mSiO2或NIP@mSiO2,充分混匀后于恒温水浴振荡器(25 ℃)吸附60 min后抽滤。上清液经0.45 μm有机系微孔滤膜过滤,通过紫外-可见分光光度计测定硝苯地平(237 nm)、尼群地平(237 nm)、苯磺酸氨氯地平(237 nm)、盐酸普萘洛尔(292 nm)的浓度,并计算吸附量。
2 实验结果与讨论
2.1 制备优化
2.1.1 功能单体用量对印迹效果的影响
功能单体主要通过氢键与目标物进行非共价结合,不同MAA用量对制备的印迹及非印迹材料吸附性能的影响见图1。MIP@mSiO2和NIP@mSiO2对硝苯地平的吸附量均随着功能单体用量增加呈现先增大后减小趋势。当模板分子/功能单体物质的量比例为1∶2时,MIP@mSiO2对硝苯地平的吸附量最大,但是印迹因子较小,可能是由于单体用量较少时,不能很好地在二氧化硅载体上形成与硝苯地平完全匹配的空间结构,难以实现对硝苯地平的特异性结合。而当模板分子/功能单体物质的量比为1∶4时,虽然MIP@mSiO2对硝苯地平的吸附量稍有下降,但是印迹因子达到最佳,为1.27。当功能单体用量继续增加时,吸附量和印迹因子均出现下降。综合考虑吸附量和特异性结合能力,选择模板与功能单体的物质的量比为1∶4进行后续优化研究。

图1 功能单体用量的影响
2.1.2 分子拥挤试剂分子量对印迹效果的影响
通过加入聚苯乙烯作为分子拥挤试剂,考察分子拥挤试剂分子量对分子印迹效果的影响。在表面印迹反应中加入分子量分别为35 000,192 000,350 000和400 000的拥挤试剂,制备的印迹聚合物吸附情况见图2。由图可见,分子拥挤试剂的加入对印迹因子的影响较为明显,与不加拥挤试剂组相比,制备的印迹聚合物吸附能力稍有下降,但印迹因子得到提升。随着拥挤试剂分子量的增大,印迹聚合物的吸附能力降低,印迹因子增大。这是因为分子拥挤环境能改变分子印迹聚合反应过程中模板分子和功能单体作用平衡,有利于聚合反应形成结构更加匹配的印迹孔穴。分子量为350 000时,吸附能力较大,印迹因子比400 000时稍低,考虑到拥挤试剂成本问题,选定分子量为350 000的分子拥挤试剂并进行后续优化。

图2 拥挤试剂分子量的影响
2.2 材料表征
2.2.1 形貌表征
如图3所示,通过扫描电镜和透射电镜对表面分子印迹材料表面形貌进行观察。从mSiO2(图3a)及MIP@mSiO2(图3b)、NIP@mSiO2(图3C)的扫描电镜图可以看出,三种材料均呈现出规则的球形颗粒状且分散性较好,载体介孔二氧化硅微球的表面较规整、光滑,粒径在550~650 nm之间;表面分子印迹微球粒径在700~800 nm之间;非印迹微球的粒径稍大,在800~900 nm之间。从MIP@mSiO2的透射电镜图(图3d)看,微球边缘有一圈半透明的表面印迹层,厚度约为50 nm。通过表面印迹聚合反应在mSiO2上涂覆了一层印迹聚合物,并且印迹反应后表面粗糙度稍有增加,表明表面分子印迹聚合成功进行。

a—mSiO2的SEM图;b—MIP@mSiO2的SEM图; c—NIP@mSiO2的SEM图;d—MIP@mSiO2的TEM图。图3 材料的扫描电镜和透射电镜表征
2.2.2 红外光谱表征
采用傅里叶变换红外光谱仪对介孔二氧化硅微球和分子印迹材料表面官能团进行分析,见图4。在mSiO2的红外光谱(图4a)中,1 069,959和794 cm-1处的峰分别归属于Si-O-Si,Si-OH以及SiO-C键的拉伸振动,3 425 cm-1处的宽峰归属于硅酸酯的-OH吸收。由图4b和4c可见,MIP@mSiO2和NIP@mSiO2的红外光谱非常相似,3 438 cm-1和1 735 cm-1处出现了强烈的吸收峰,这是来自于羧基O-H的伸缩振动和聚甲基丙烯酸酯C=O的伸缩振动,在2 985 cm-1和2 952 cm-1处吸收峰归因于-CH3和-CH2-的不对称伸缩振动。傅立叶红外光谱表明,表面印迹层已成功涂覆于二氧化硅微球载体上。
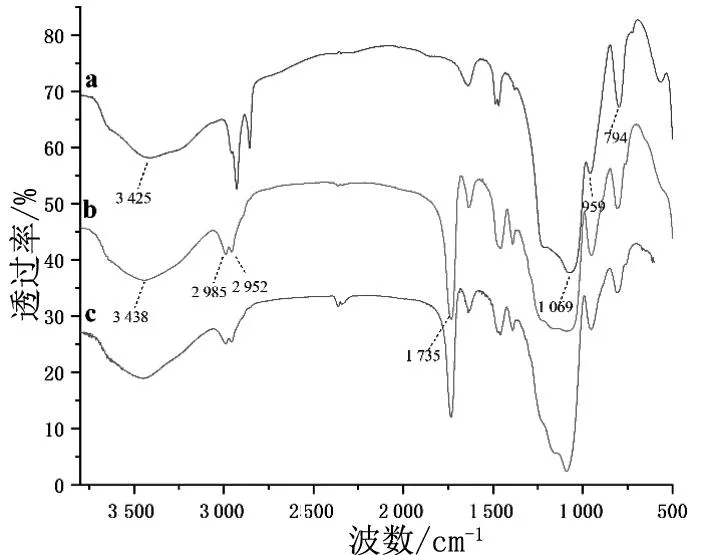
a—mSiO2;b—MIP@mSiO2;c—NIP@mSiO2。图4 材料的红外光谱
2.3 吸附性能
2.3.1 饱和吸附性能考察
考察了MIP@mSiO2和NIP@mSiO2材料在系列质量浓度(5~150 mg/L)的待吸附液中的静态吸附性能,见图5。在质量浓度为5~75 mg/L时,MIP@mSiO2和NIP@mSiO2材料对硝苯地平的吸附量都随着待吸附母液中硝苯地平浓度的增大而增大。当质量浓度为100 mg/L时,两种材料的吸附量逐渐趋于平衡,分别为153.1 mg/g和107.2 mg/g。这是因为印迹材料表面存在与目标分子结构匹配的印迹孔穴,而非印迹材料没有,导致印迹材料的吸附能力比非印迹材料大。
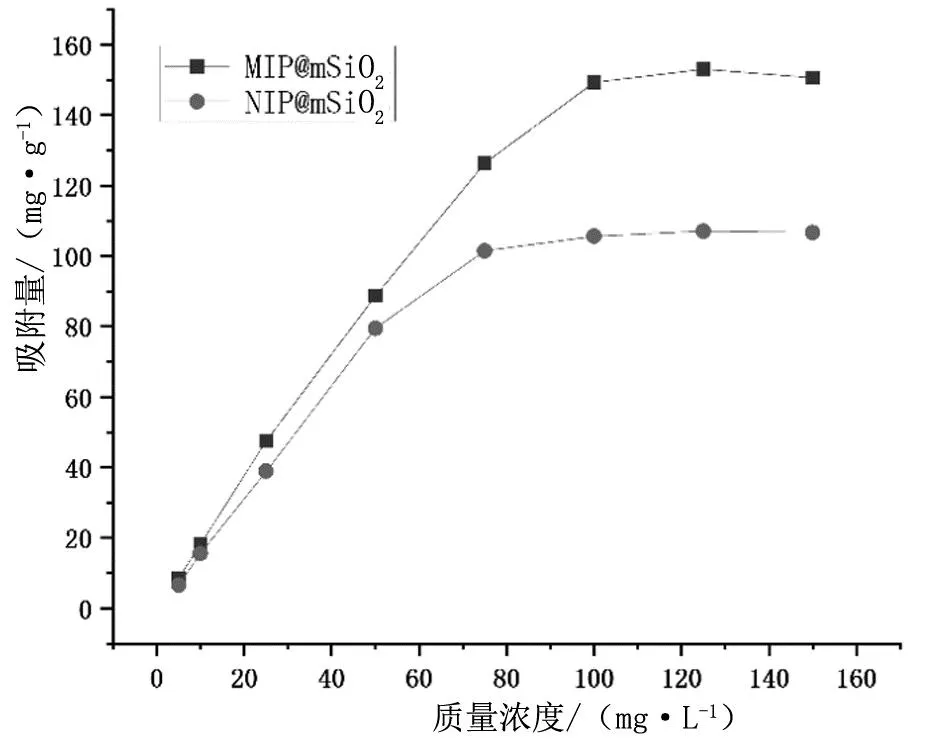
图5 饱和吸附性能曲线
2.3.2 吸附动力学
按1.3.2考察MIP@mSiO2和NIP@mSiO2在不同的吸附时间取样时计算的吸附能力。以吸附取样时间作为横坐标,吸附量作为纵坐标,得到表面印迹微球的吸附动力学曲线,见图6。由图可知,MIP@mSiO2对硝苯地平在20 min内已趋于饱和吸附,30 min时达到饱和吸附,吸附量约为149.6 mg/g;NIP@mSiO2对硝苯地平吸附在45 min内达到饱和吸附,饱和吸附量约为103.4 mg/g。这是由于表面分子印迹材料表面存在大量的印迹孔穴,对硝苯地平分子具有较快的传质速率,具有较快速的吸附动力学;而随着表面印迹孔穴逐渐被硝苯地平分子占据,吸附趋于饱和。非印迹材料对目标物的吸附仅仅依靠表面的非共价作用,吸附速率相对较慢。
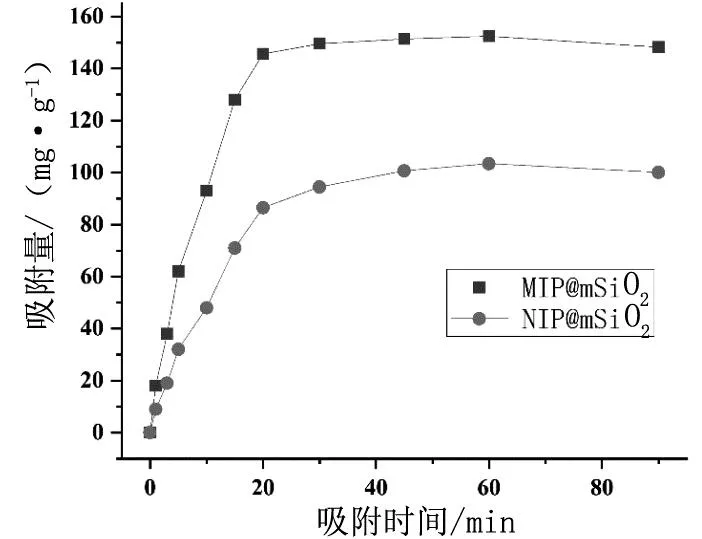
图6 吸附动力学曲线
2.3.3 选择性吸附
选择与硝苯地平结构相似的尼群地平、苯磺酸氨氯地平和结构不同的盐酸普萘洛尔,用于评估MIP@mSiO2和NIP@mSiO2的吸附选择性。由图7可知,MIP@mSiO2对模板硝苯地平的吸附量最高,而对尼群地平、苯磺酸氨氯地平和盐酸普萘洛尔的吸附量较低,特别是对结构不同的盐酸普萘洛尔的吸附量最低;而NIP@mSiO2对四种底物的吸附量均较低。这是因为分子印迹材料表面的印迹孔穴在结合位点的官能团和三维立体结构与模板分子硝苯地平匹配性高,吸附选择性更好。

图7 材料的选择性吸附
2.3.4 循环使用性能
为探究MIP@mSiO2使用的可重用性和稳定性,将吸附后的MIP@mSiO2用甲醇/乙酸(体积比9∶1)溶液洗脱多次,除去模板,再用甲醇洗涤两次,在50 ℃下干燥至恒重,进行下一周期的静态吸附测试。如此循环多次并计算每次的吸附量。图8显示经过3次吸附-脱附循环后,MIP@mSiO2对硝苯地平的静态吸附能力保持94%以上,且5个吸附周期后,表面印迹材料的静态吸附能力只有略微的损失。这表明制备的表面分子印迹材料稳定性较好,具有一定可循环使用价值。

图8 材料的循环使用性能
3 结论
(1)采用表面印迹技术和分子拥挤技术,以硝苯地平为模板分子,制备了表面分子印迹聚合微球材料。
(2)表面分子印迹微球的粒径在700~800 nm之间,印迹层厚度约为50 nm。
(3)分子拥挤试剂的加入对印迹因子的影响较为明显,与不加拥挤试剂组相比,制备的印迹聚合物吸附能力稍有下降,但印迹因子得到提升。
(4)表面分子印迹聚合材料具有较大的饱和吸附量(153.1 mg/g)和快速的吸附动力学(30 min),材料对结构相似的竞争物具有较好的选择性,3次吸附-脱附循环使用吸附性能仍保持94%以上。
