Ni1/g-C3N4催化乙炔加氢反应的密度泛函理论研究
2024-01-02侯翠丽康丽华李洪玲
侯翠丽 康丽华 李洪玲


摘要:乙烯是石化产业的主要原料之一,广泛运用于工业生产和基本生活。乙烯中少量的乙炔杂质从根本上会影响下游产品,那么乙炔选择性加氢就可以提高乙烯产量,减少杂质产生。本文中构建了Ni1/g-C3N4催化剂模型,以密度泛函理论为依据,从B3LYP泛函基组出发,计算过程中采用DFT-D3校正。对Ni原子使用LAN2DZ赝势,对于C, H, N非金属原子使用6-31g**基组。通过分析静电势、态密度和在催化剂上的吸附行为,系统性研究了Ni1/g-C3N4催化剂催化乙炔加氢的反应机理,详细阐述了选择性和活性。结果表明,Ni1/g-C3N4催化乙炔加氢的最优反应路径是乙炔加氢生成乙烯,能垒是20.05 kcal·mol-1;乙烯加氢生成乙烷,能垒是90.29 kcal·mol-1。Ni1/g-C3N4催化剂具有低活性、高选择性的特征,展现了其良好的催化性能,为乙炔高效催化加氢领域提供了帮助。
关键词:乙炔加氢;密度泛函理论;单原子催化剂;活性;选择性
中图分类号:TQ221.211;TQ426中图分类号文献标志码:A文献标识码
Density functional theory study on Ni1/g-C3N4 catalytic hydrogenation
of acetylene
HOU Cuili,KANG Lihua,LI Hongling*
(School of Chemistry and Chemical Engineering, Shihezi University, Shihezi,Xinjiang 832003, China)
Abstract: Ethylene is one of the main raw materials in the petrochemical industry and is widely used in industrial production and basic life. A small amount of acetylene impurities in ethylene will fundamentally affect downstream products, so selective hydrogenation of acetylene can increase ethylene production and reduce impurities. In this paper, the Ni1/g-C3N4 catalyst model is constructed, based on density functional theory, starting from the B3LYP functional basis set, and using DFT-D3 correction in the calculation process. The LAN2DZ pseudopotential was used for Ni atoms and the 6-31g** basis set for C, H, N non-metallic atoms. By analyzing the electrostatic potential, density of states and adsorption behavior on the catalyst, the reaction mechanism of Ni1/g-C3N4 catalyst for acetylene hydrogenation was systematically studied, and the selectivity and activity were also elaborated. The results show that the optimal reaction path for Ni1/g-C3N4 catalyzed acetylene hydrogenation is hydrogenation of acetylene to ethylene with an energy barrier of 20.05 kcal·mol-1; hydrogenation of ethylene to ethane with an energy barrier of 90.29 kcal·mol-1. The Ni1/g-C3N4 catalyst has the characteristics of low activity and high selectivity, showing its good catalytic performance, which provides help for the field of efficient catalytic hydrogenation of acetylene.
Key words: Acetylene hydrogenation;density functional theory;single-atom catalysts;activity;selectivity
在现代化工业中,乙烯是石油化工产品中的主流原料和基本合成单体[1]。石油烃类在经过蒸汽裂解得到乙烯的过程中会产生痕量乙炔,这些乙炔在乙烯后续的反应特别是乙烯聚合过程中,破坏乙烯的聚合,导致催化剂失活,进一步降低催化剂的选择性和使用寿命[2-3]。传统的乙炔加氢工艺复杂、操作严格、污染环境,并不能作为工业上的常用方法。由于在反应过程中乙烷是热力学更稳定的结构,因此乙炔加氢所产生的乙烯以及原料气中的乙烯很容易被过度加氢到乙烷。乙烯进一步受到损耗,因此在乙炔选择性加氢反应中如何控制反应目标产物乙烯不进一步发生加氢反应是技术难点。在社会经济的高速发展和能源紧缺问题日益严重的今天,开发一种具有优异催化性能的催化剂势在必行。有研究表明,基于泡沫玻璃原理构建的结构催化剂在催化加氢方面得到迅速发展,王嵩[4]以泡沫镍为载体通过等体积浸渍法制备了Pd/Ni-foam结构催化剂,通过实验表明镍基类催化剂在乙炔加氢的过程中表現出了高乙烯选择性,但乙炔转化成乙烯的转化率较低。为了提高镍基催化剂的性能,单原子催化剂进行催化加氢的概念被提出,这种利用金属Ni活性中心进行有效加氢的方式引起反响[5-8]。Dai等[9]根据以上方法进行了大量的试验,提出了单个金属Ni催化剂用于乙炔加氢时,展现出了巨大的加氢潜力。Chai等[10]通过实验表明限制在沸石中的四配位阳离子镍 (II) 能够有效地催化乙炔选择性氢化成乙烯,对去除痕量乙炔起到了关键作用,在优化时可同时实现 100% 的乙炔转化率和高达 97% 的乙烯选择性。
据最近研究,石墨化氮化碳(g-C3N4)被广泛用作催化载体[11-12]。Li等[13]制备的超薄g-C3N4纳米片具有比表面积大,更多的催化活性位点等特点。同时,Pd基在乙炔催化加氢方面表现出优异的活性和选择性。但Pd作为贵金属价格昂贵,因此我们选取与Pd在同一副族的非贵金属Ni。我们猜测两者存在类似的催化性能,但到目前为止还没有关于单金属Ni1/g-C3N4催化剂的理论计算报道[14-15]。因此,本文选取g-C3N4为载体,将Ni原子吸附于g-C3N4六空腔中,优化并得到Ni1/g-C3N4最稳定的结构,探究Ni1/g-C3N4催化剂催化乙炔加氢的反应路线和机理,对反应的活性和选择性进行了计算和总结。总之,此次探究为单金属Ni和二维层状材料结合进行催化反应领域拓宽了研究方向。
1 计算方法
本研究运用的DFT计算过程均在Guassian09程序软件包完成[16]。Tentscher等[17]对于不同的泛函进行了测试,结果表明B3LYP[18]混合密度泛函理论可以适用于反应过程的几何优化。Yang等[19]表明使用B3LYP泛函时,6-31g**和LANL2DZ构成的混合基组具有较好的性能。基于整个反应路径中的几何优化和过渡态,6-31G**基组被应用于 C、N、H 原子,金属Ni原子运用LANL2DZ基组。在反应路径中,所有进行优化的结构均考虑了Grimme经验色散矫正,其优化结构的相对能量也由相等水平下的零点能量校正计算得出。在反应进行过程中,在几何优化过程中不存在对称约束,频率计算能够确保每一个过渡态都仅有一个虚频,也可以用于查验所有不同的稳定点是否为最小值。在计算进行中,利用本征反应坐标(IRC)[20]可以确保过渡态连接的产物和反应物是正确的。另外,计算了C2H2和H2在Ni1/g-C3N4催化剂上的吸附能,将单吸附能(Eads)和共吸附能(Eco-ads)定义为:
其中,E乙烷生成表示生成乙烷的能垒,E乙烯脱附表示乙烯脱附时的能垒。
2 结果与讨论
2.1 催化剂的结构与性质
2.1.1 催化剂模型的构建及优化
大量研究表明,g-C3N4存在两种基本的结构,即为三嗪(C3N4)和3-s-三嗪(C6N7),通過理论计算进行研究和探索,结果显示3-s-三嗪(C6N7)比三嗪(C3N4)较稳定[21-22]。由于3-s-三嗪结构的每个基本单元外围都存在一个活性N原子,并且这些边缘活性N原子具有可连性。因此可利用这种特性,选择其中3个基本单元将其连接围成一个最佳的活性空间,进一步找到最优活性位点。为探究最大活性空间的存在位置,本次理论研究中,选择结构为3个相邻片状的3-s-三嗪(C6N7)单元作为主体,对其进行建模并优化。其结构如图1所示。
为了得到Ni1/g-C3N4最优且最稳定的结构,我们将 Ni原子放置在图1a中的不同位置。如 C1, N1, N2的顶位,N1-N2, N2-N3, N3-N4 的桥位,六空穴位处以及所有可能存在的位置,结果表明Ni原子都倾向于吸附在g-C3N4的六空腔中,其结构分别如图1b所示。其中,Ni原子大致位于g-C3N4六空腔的中间,且Ni原子与 C1 原子相连成键,键长为1.93 。Ni-N1、Ni-N2、Ni-N3、Ni-N4、Ni-N5、Ni-N6的距离分别为2.36 、2.17、2.84、3.34、2.69、2.06 。从图中看出,Ni原子吸附到g-C3N4上之后,催化剂发生形变,尾部向上轻微翘起,表明金属原子Ni和载体g-C3N4之间存在强烈的相互作用。反应物C2H2和H2分子优化后的构型如图1中展示,直链C2H2分子C≡C键长为1.21 ,H2分子的H-H键长为0.74 。
2.1.2 催化剂的电子性质
为了进一步了解电荷分布情况,绘制了范德华表面的静电势图(电子密度为 0.001),分别为吸附金属原子前催化剂载体g-C3N4和吸附金属原子后催化剂Ni1/g-C3N4。蓝色或红色的颜色越深,表示正电荷或负电荷越密集。如图2所示,吸附金属原子前催化剂载体g-C3N4六空腔中负电荷较为密集,但六空腔中央未有电子存在。然而,吸附金属Ni原子后,六空腔中空缺电子位置被充满,Ni原子周围负电荷分布十分充裕。因此,根据催化剂表面的分子静电势图可推测,金属原子Ni是整个吸附反应的活性位点。
为了更加深入了解g-C3N4吸附金属原子前后的特征,将分子轨道变化和各组分对总态密度的贡献作为主要分析对象。如图3所示,通过Multiwfn软件[23]绘制g-C3N4和Ni1/g-C3N4的TDOS图,以及相应的HOMO、LUMO轨道。首先观察态密度图能够发现,在g-C3N4中,HOMO的主要贡献轨道是N原子的p轨道,C原子的p轨道和N原子的p轨道分别是LUMO的主、次要贡献轨道。吸附金属Ni原子后,HOMO轨道中各组分的贡献发生了变化,对LUMO轨道的贡献没有影响。C原子的p轨道和N原子的p轨道仍为LUMO的主、次要贡献轨道,但HOMO的主要贡献轨道变成了Ni原子的d轨道,次要贡献轨道是N原子的p轨道和C原子的p轨道。通过HOMO图能够看出,g-C3N4中的电荷分布相对均匀,而吸附金属原子之后,电荷主要集中在金属Ni原子周围。
结果表明金属Ni原子和g-C3N4载体两者之间存在明显的电荷转移,金属Ni原子是反应的活性位点。此外,在元素周期表中,Ni原子的价电子排布为3d84s2,和态密度图相结合很明显看出Ni原子处的 “d”轨道电子呈现未充满状态。
2.2 催化剂上反应物的吸附行为
对g-C3N4和Ni1/g-C3N4特征进行了以上初步分析后,猜想反应的活性位点是金属原子Ni。在进行吸附反应时,需要明确反应的活性位点以及反应物C2H2和H2的先后吸附顺序。将反应物C2H2和H2分别吸附于Ni1/g-C3N4催化剂上,图4展示了单吸附最稳定的结构。如图4a显示,C2H2吸附在催化剂上后,C2H2分子从原来的C≡C变成了CC双键,C1和C2均与金属Ni相连,C1和C2之间的键长也从1.20 增加至1.27 。随之C2H2分子也发生相应的形变,说明C2H2与Ni1/g-C3N4存在一定的相互作用。从图4b显示,H2吸附在催化剂上之后,金属原子Ni与H原子相连,且H-Ni键长从0.79 增加至1.53 。
接下来,利用电荷密度差分图(EDD)(电子密度为0.001 a.u.)对其进一步分析,如图5所示。从图5中可以看出,反应物C2H2和H2与催化剂Ni1/g-C3N4均存在相互作用,且C2H2表现出更强的相互作用。由此可说明金属原子Ni就是唯一的活性位点,这也与前文分析的静电势以及态密度图结果一致。通过表1单吸附和共吸附能明显看出,C2H2的吸附能大于H2,表明在吸附时,C2H2更容易优先吸附,再后吸附H2,最后形成共吸附。另外,C2H2和H2的单吸附能均小于其对应的共吸附能,这确保了进行反应时可以在合理范围内从单吸附结构转化为共吸附结构。
2.3 Ni-C3N4催化乙炔加氢反应
为了更好的研究 Ni1/g-C3N4催化乙炔加氢的
具体反应路径,在寻找C2H2和H2在催化剂表面进行共吸附时,发现了R1和R2两种不同的共吸附结构。立足于乙炔加氢的反应机理,将反应路径大致分为乙炔一次加氢生成乙烯和二次加氢生成乙烷两个部分。对在催化剂表面进行的两个可能的反应路径做了全面的摸索和分析,通过对比得出了最优反应路径,并对其反应的活性和选择性做了详细的阐述。
2.3.1 Ni1/g-C3N4催化乙炔加氢的反应路径一
Ni1/g-C3N4催化乙炔加氢反应的第一条路径从共吸附 R1开始,乙炔催化生成乙烯的反应路径及其每一步对应的能量如图6所示。在共吸附R1 中,C2H2 和 H2 处在Ni1/g-C3N4的同一个平面上,并且乙炔在氢气分子的左方位,该共吸附的共吸附能为-41.88 kcal·mol-1。从共吸附R1开始到生成乙烯IM3,需要经过3个过渡态TS1、TS2、TS3,并且存在两个中间体IM1和IM2。首先,共吸附R1需要越过能垒为 20.05 kcal·mol-1的过渡态 TS1 才能生成中间体 IM1,该过程称为速率决定步骤。在 TS1 中,C2H2 分子翻转到Ni原子的左侧,相应的Ni原子随之也扭转到C2H2 分子的右侧,两个 H 原子同在右侧,H1-H2 键长从 0.81 伸长到 2.19 ,表明H2分子已被完全活化。H1 原子朝 C1 原子振动,并且只有一个值为-843.67 cm-1的虚频。在 IM1 中,H1 和 C1键长从 2.30 缩短至 1.18 并成键,由于 H2 原子离 C2 原子较远,因此IM1生成乙烯IM3需要经历两个过渡态。在TS2 中,H2 原子朝 C2 原子振动,Ni-C2-C1 的键角从 83.20°扩大到 107.77°,过程中需要越过7.34 kcal·mol-1的能垒生成中间体 IM2,且只有一个-668.81cm-1的虚频。在 IM2 中,C2原子与 H2 原子离的较近,只需要克服 2.02 kcal·mol-1的能垒生成乙烯。在 TS3 中,Ni-C2-C1的键角从78.41°缩小到57.35°,且仅得到了一个-709.81 cm-1的虚频。在IM3中,C2-H2的键长从2.07 缩短至1.14 并成键产生乙烯,这是乙炔加氢的第一步产物。此时,乙烯的脱附能为39.05 kcal·mol-1。在这个过程中,使用 IRC 验证并确保了所有过渡态的准确性,即每一个过渡态都印照着相应的反应物和中间体。
为了研究反应的选择性,也深层次的研究了乙烯的进一步加氢的反应路径。
首先,将H2 分子置于乙烯附近可能的位置,并得到了最优的共吸附结构(IM4)。其次,C2H2 和 H2 仍然处于Ni原子两侧的平面,但与乙炔加氢時有所不同,在乙烯加氢生成乙烷的反应过程中,仅需要2个过渡态(TS4和TS5)就可生成产物乙烷。IM4越过90.29 kcal·mol-1的能垒经历过渡态 TS4形成中间体 IM5,该加氢过程是乙烯加氢生成乙烷的速率决定步骤。在 TS4 中,H3原子向C1原子振动,该过渡态仅有1个-1415.38 cm-1的虚频。在 IM5 中,C1-H3 间距缩短至1.09 后成键,H4 原子与Ni原子成键,键长为1.45 。随后,H4 原子与Ni原子的成键断裂,并至少需要4.74 kcal·mol-1能垒经过过渡态TS5 生成乙烷(P)。
在TS5中,H4 原子朝 C2 原子振动且只有1个-236.10 cm-1的虚频,H4-C2的键长从为2.36 急剧缩短至1.10 。经过 IRC的计算,验证了每一步反应中的过渡态都正确地与反应前后的结构相连,并且过程中无其他中间体(图7)。
2.3.2 Ni1/g-C3N4催化乙炔加氢的反应路径二
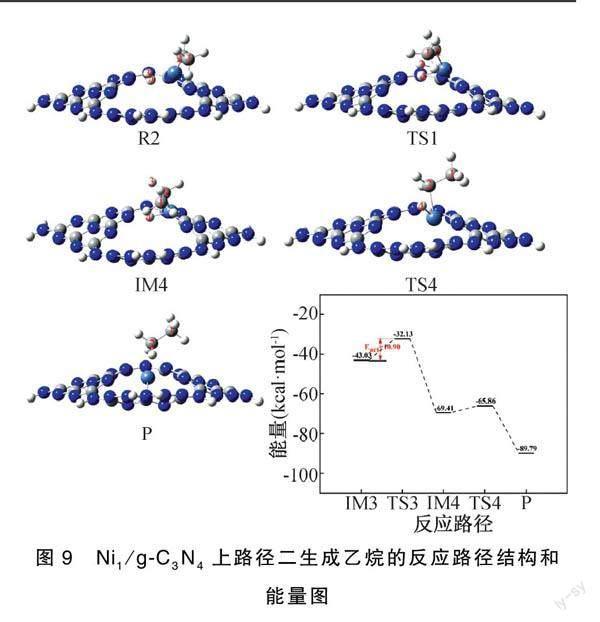
首先,Ni1/g-C3N4 催化乙炔加氢反应的第二条路径从共吸附R2开始,将会经过TS1、TS2两个过渡态和中间体IM1生成次乙基(IM7)。在R2共吸附中,C1和C2处于H原子右侧,大致在一个平面,共吸附能为-26.49 kcal·mol-1。R2 生成中间体IM1需要克服40.07 kcal·mol-1的能垒并经过过渡态 TS1才能形成。在TS6中,同时H1和H2原子逐渐靠近C1和C2,说明H1和H2已经活化。在IM1中,H1-C2键长缩短至1.10 并成键,H2-Ni键长缩短至1.43 并成键。生成次乙基 IM2需要越过40.51 kcal·mol-1的能垒通过过渡态TS2,该步骤速率控制步。在TS2中,H2向C1振动,H2-C1键长从2.63 缩短到1.13 。此时,C1-C2-Ni键角为91.26°。在IM2中,H2-C1相连时键长为1.09 ,C1-C2双键断裂,C1-C2-Ni角度扩大至123.24°,键角逐渐趋于平缓。TS1和TS2的虚频分别为-720.84 cm-1和-720.84 cm-1,均只有一个虚频(图8)。
其次,为探究生成乙烷的反应,IM2将会二次加氢生成产物乙烷,H2被放在次乙基附近的不同位置后,选择最优的吸附结构(IM3)。随后IM3将克服10.90 kcal·mol-1的能垒经过TS3生成IM4。在TS3中,对TS3振动分析得到唯一的虚频(-990.40 cm-1),其振动表现为H3、H4键长从0.81 扩大到1.83 ;H3原子朝着靠近C2原子的方向振动。H3-C2的键长从2.70 缩短到1.14 。在生成IM4时,H3原子逐渐移动到C2原子的左上方并与之相连,H3-C2的键长为1.09 。紧接着IM4将要越过3.55 kcal·mol-1的能垒并经过过渡态TS4生成最终的产物乙烷(P)。在TS4 中,随着H4-Ni-C2键角从82.32°缩小到36.40°时,H4-C2的键长缩短到1.12,且只有一个虚频为-709.81 cm-1。生成产物乙烷后,Ni原子也解析到催化剂g-C3N4上。IRC计算并验证了过渡态的正确性,其结果证明连接了反应前后的结构,过程中无其他中间体(图9)。
2.3.3 Ni1/g-C3N4的活性和选择性
第一,我们来讨论Ni1/g-C3N4的活性。根据上文探究发现:金属Ni原子加入g-C3N4,Ni原子是唯一支撑活性位点,增加了Ni1/g-C3N4催化剂的活性。在反应过程中,催化剂Ni1/g-C3N4具有较低的生成乙烯的能垒,证实了乙炔加氢过程中Ni1/g-C3N4催化剂表现出优异的活性。表2中罗列出一些前人探索有关催化剂催化乙炔加氢反应的对比结果,Ni1/g-C3N4催化剂具有低能垒和高活性的特征,进一步说明Ni1/g-C3N4的催化性能较好。
第二,对Ni1/g-C3N4的选择性而言,判断选择性存在两种方式。其一是乙炔加氢生成乙烯的活化能(20.05 kcal·mol-1)低于乙烯加氢生成乙烷的活化能(90.29 kcal·mol-1),两者差值越大,表明Ni1/g-C3N4选择性较高。其二是计算法,即乙烯的选择性等于乙烯加氢生成乙烷的活化能减去乙烯的脱附能,我们可以得到乙烯的选择性为51.24。从表2可以看出,Ni1/g-C3N4具有高选择性。它意味着在催化乙炔加氢的反应过程中,总是更容易向生成乙烯的方向发生,没有副产物的影响。
此外,我们在Ni1/g-C3N4催化剂催化乙炔加氢的整个反应路径中,选取不同的泛函(基组)进行比较,如图10所示。测试结果表明,使用不同的泛函或使用大基组对催化加氢的反应过程的模拟结果影响相对较小。
3 结论
(1) 乙炔加氢的最佳反应路径为由R1开始乙炔的连续性加氢的路径,分别为乙炔一次加氢生成乙烯,和乙炔二次加氢由乙烯产生乙烷。能垒分别为20.05 kcal·mol-1和90.29 kcal·mol-1,选择性为51.24。
(2) 由R2开始生成乙烷的路径能垒较高,选择性较差,不利于催化剂催化乙炔加氢。
(3) 通过与其他乙炔加氢的催化剂进行对比,Ni1/g-C3N4催化剂表现出低能垒、选择性高的催化性能,是乙炔加氢潜在的催化剂。
参考文献(References)
[1]黄伟民,周静,徐逸琦,等.乙炔选择性加氢催化剂的研究进展[J].高校化学工程学报,2020,34(6):1339-1353.
HUANG W M,ZHOU J,XU Y Q,et al.Research progress on catalysts for selective hydrogenation of acetylene[J].Journal of Chemical Engineering in Universities,2020,34(6):1339-1353.
[2]BORODZINSKI A,BOND G C.Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts,Part 2:Steady-state kinetics and effects of palladium particle size,carbon monoxide,and promoters[J].Catalysis Reviews-Science and Engineering,2008,50(3):379-469.
[3]BORODZINSKI A.Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts Part 1.Effect of changes to the catalyst during reaction[J].Catalysis Reviews-Science and Engineering,2006,48(2):91-144.
[4]王嵩.整裝金属纤维/泡沫结构化 Pd,Ni 基催化剂及其乙炔选择性加氢催化性能研究[D].上海:华东师范大学,2020.
WANG S.Monolithic metal fiber-/foam-structured Pd-/Ni based catalysts for selective hydrogenation of acetylene to ethylene[D].East China Normal University,2020.
[5]WANG Y W,SONG E H,QIU W J,et al.Recent progress in theoretical and computational investigations of structural stability and activity of single-atom electrocatalysts[J].Progress in Natural Science-Materials International,2019,29(3):256-264.
[6]GIANNAKAKIS G,FLYTZANIl M,SYKES E C H.Single-atom alloys as a reductionist approach to the rational design of heterogeneous catalysts[J].Accounts of Chemical Research,2019,52(1):237-247.
[7]WANG A Q,LI J,ZHANG T.Heterogeneous single-atom catalysis[J].Nature Reviews Chemistry,2018,2(6):65-81.
[8]LIU J Y.Catalysis by supported single metal atoms[J].ACS Catalysis,2017,7(1):34-59.
[9]DAI X Y,CHEN Z,YAO T,et al.Single Ni sites distributed on N-doped carbon for selective hydrogenation of acetylene[J].Chemical Communications,2017,53(84):11568-11571.
[10]CHAI Y C,WU G J,LIU X Y,et al.Acetylene-selective hydrogenation catalyzed by cationic confined in zeolite[J].Journal of the American Chemical Society,2019,141(25):9920-9927.
[11]CHEN Z,ZHAO J X,CABRERA C R,et al.Computational screening of efficient single-atom catalysts based on graphitic carbon nitride (g-C3N4) for nitrogen electroreduction[J].Small Methods,2019,3(6):1800368.
[12]ZHANG L K,ZHANG L H,CHENG S S,et al.Pd supported on graphene modified g-C3N4 hybrid:a highly efficient catalyst for hydrogenation of nitroarenes[J].Applied Organometallic Chemistry,2020,34(8).
[12]REN Y J,ZENG D Q,ONG W J.Interfacial engineering of graphitic carbon nitride (g-C3N4)-based metal sulfide heterojunction photocatalysts for energy conversion:A review[J].Chinese Journal of Catalysis,2019,40(3):289-319.
[13]LI Y F,JIN R X,XING Y,et al.Macroscopic foam-like holey ultrathin g-C3N4 nanosheets for drastic improvement of visible-light photocatalytic activity[J].Advanced Energy Materials,2016,6(24):16012731-16012735.
[14]LIU H,CHAI M Q,PEI G X,et al.Effect of IB-metal on Ni/SiO2 catalyst for selective hydrogenation of acetylene[J].Chinese Journal of Catalysis,2020,41(7):1099-1108.
[15]HEARD C J,HU C Q,SKOGLUNDH M,et al.Kinetic regimes in ethylene hydrogenation over transition-metal surfaces[J].ACS Catalysis,2016,6(5):3277-3286.
[16]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09 Citation[B/OL].Available online:https://gaussian.com/g09citation/.Gaussian Inc.:Wa-llingford,CT,USA,2010.
[16]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09,Revision B.01[J].2010.
[17]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09 (Revision D.01)[J].2009.
[17]TENTSCHER P R,AREY J S.Geometries and vibrational frequencies of small radicals:performance of coupled cluster and more approximate methods[J].Journal of Chemical Theory and Computation,2012,8(6):2165-2179.
[18]LEE C,YANG W,PARR R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Physical Review B,1988,37(2):785-789.
[19]YANG Y,WEAVER M N,MERZ K M.Assessment of the “6-31+G** + LANL2DZ” mixed basis set coupled with density functional theory methods and the effective core potential:prediction of heats of formation and ionization potentials for first-row-transition-metal complexes[J].The Journal of Physical Chemistry A,2009,113(36):9843-9851.
[20]GONZALEZ C,SCHLEGEL H B.Reaction-path following in mass-weighted internal coordinates[J].The Journal of Physical Chemistry,1990,94(14):5523-5527.
[21]ALVES I,DEMAZEAU G,TANGUY B,et al.On a new model of the graphitic form of C3N4[J].Solid State Communications,1999,109(11):697-701.
[22]MIYAMOTO Y,COHEN M L,LOUIE S G.Theoretical investigation of graphitic carbon nitride and possible tubule forms[J].Solid State Communications,1997,102(8):605-608.
[23]LU T,CHEN F W.Multiwfn:A multifunctional wavefunction analyzer[J].Journal of Computational Chemistry,2012,33(5):580-592.
[24]WANG Y,KANG L H.Selective hydrogenation of acetylene catalyzed by nickel and nitrogen-doped C34:A density functional theory study[J].Chemical Physics Letters,2020,757:137871.
[25]YANG B,BURCH R,HARDACRE C,et al.Origin of the increase of activity and selectivity of nickel doped by Au,Ag,and Cu for acetylene hydrogenation[J].ACS Catalysis,2012,2(6):1027-1032.
[26]ZHAO Y,ZHU M Y,KANG L H.The DFT Study of Single-Atom Pd-1/g-C3N4 catalyst for selective acetylene hydrogenation reaction[J].Catalysis Letters,2018,148(10):2992-3002.
[27]WANG Y,KANG L H.Selective hydrogenation of acetylene catalysed by a B12N12 cluster doped with a single nickel atom:a dft study[J].Catalysts,2020,10(1):115.
(責任编辑:编辑唐慧)
收稿日期:中文收稿日期2022-05-03
基金项目:国家自然科学基金项目(51563021)
作者简介:侯翠丽(1996—),女,硕士研究生,专业方向为化学。
*通信作者: 李洪玲(1971—),女,教授,从事化学工艺研究,e-mail: lihongling197102@163.com。
