咖啡中3种赭曲霉毒素QuEChERS-UPLC-MS/MS检测方法
2023-12-28高云慨陈小妹陈春泉周凌聿邓英林尹青春
高云慨 陈小妹 陈春泉 周凌聿 邓英林 尹青春
(海南省食品检验检测中心国家市场监管重点实验室〔热带果蔬质量与安全〕,海南 海口 570100)
据统计[1],中国饮用咖啡的消费者已达3.3亿人,近年来咖啡消费市场规模保持20%的年化速度飞速增长。中国咖啡种植主要分布在温润潮湿的云南、四川及海南地区。研究[2]表明,咖啡及其产品在采收、加工、贮藏过程中易受到霉菌的侵染,产生各种真菌毒素。Bessaire等[3]采集了9个国家咖啡样本,多数样本中含有多种霉菌毒素,其中赭曲霉毒素A含量最高。饮用咖啡是机体摄入该毒素的主要途径,因在生产及食用环节难以完全避免其毒性,已成为危害健康的主要关键风险因素[4-5]。
赭曲霉毒素(ochratoxin,OT)是由真菌产生的具有结构类似的次生代谢产物,常见的有赭曲霉毒素A(OTA)、赭曲霉毒素B(OTB)和赭曲霉毒素C(OTC)等[6-7]。其主要通过侵害动物肝脏与肾脏,具有致癌、致畸等毒副作用,已被国际癌症研究机构(IARC)列入2B类致癌目录,危害性仅次于黄曲霉毒素[8]。研究[9]表明,OTA和OTC在特定条件下可以相互转化,具有协同毒副效应。同时,代谢的多种真菌毒素的协同作用对人体健康的影响更具危害性[10-11]。叶林链等[12]从肉豆蔻中分离了一株赭曲霉毒素产毒菌,同时检出了OTA、OTB两种真菌毒素,说明在植物源样本中存在不同结构的赭曲霉毒素。GB 2761—2017对咖啡中赭曲霉毒素A限值有明确要求,但其他赭曲霉毒素类型尚未见相关规定[13]。
近年来,高效液相色谱—串联质谱技术因其强大的分离能力和较好的灵敏度、准确性,逐步被应用于部分真菌毒素的定性和定量分析[14-18]。免疫亲和柱法对真菌毒素具有高特异性,是目前国家标准检测咖啡赭曲霉毒素A采用的前处理方法,但由于采用抗原抗体结合的净化方式,其前处理过程复杂、使用成本高,检测的真菌毒素种类有限。相比免疫亲和柱,QuEChERS前处理技术采用提取与净化结合方式,具有操作简单、快速、价格便宜且选择性好等优点,可应用于多种真菌毒素的高通量检测[19-23]。
目前国内有关咖啡及制品中真菌毒素检测的研究较少[24],尚未见针对咖啡中同时检测OTA、OTB、OTC 3种类型赭曲霉毒素方法研究及评价的报道。研究拟将高效液相色谱—串联质谱技术与QuEChERS技术结合,采用内标法定量,旨在建立能快速筛查和准确定量咖啡中3种赭曲霉毒素检测方法,以期为全面监测、评估咖啡中赭曲霉毒素的污染风险提供依据。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
OTA、OTC:天津阿尔塔科技有限公司;
OTB:德国Dr. Ehrenstorfer公司;
13C20-OTA:北京曼哈格生物科技有限公司;
QuEChERS提取盐包:4 g硫酸镁、1.0 g氯化钠,美国Agilent公司;
净化柱:SHIMADZU WondaPak QuEChERS净化管,日本SHIMADZU公司;
Waters Oasis®PRime HLB:200 mg,6 mL,美国Waters公司;
ZanChERS-Myco17净化小柱:北京科德诺思技术有限公司;
色谱柱:ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm),美国Waters公司;
乙腈、甲酸、甲醇:德国MERCK公司;
咖啡生豆、烘焙咖啡豆、速溶咖啡粉:市售。
1.1.2 仪器与设备
质谱仪:AB Sciex Triple QuadTM4500型,美国SCTEX公司;
电子天平:XS204S型,瑞士梅特勒—托利多公司;
冷冻离心机:5804R型,德国Eppendorf公司;
超声波器:SK 7200型,上海科导超声仪器有限公司。
1.2 方法
1.2.1 仪器条件
(1) 质谱条件:离子源(ESI);正离子模式;多反应监测(MRM)扫描;喷雾电压4 500 V;离子源温度550 ℃;雾化气为氮气;加热气为氮气;碰撞室入口电压10 V。
(2) 色谱条件:ACQUITY UPLC BEH C18色谱柱(2.1 mm×100 mm,1.7 μm);柱温40 ℃;进样体积5.0 μL;流动相A为0.1%甲酸水溶液;流动相B为乙腈;流速0.3 mL/min;洗脱程序:0~2.0 min,10% B;2.0~3.0 min,10%~90% B;3.0~3.2 min,90%~10% B;3.2~5.0 min,10% B。
1.2.2 标准溶液配制
(1) 混合标准溶液(1.0 μg/mL):准确吸取100 μg/mL 的3种赭曲霉毒素混标100 μL,用乙腈定容至10 mL,并于-18 ℃贮藏备用。
(2)13C20-OTA内标使用溶液(10.0 μg/mL):准确吸取100 μg/mL13C20-OTA溶液1.0 mL,用乙腈定容至10 mL,并于-18 ℃贮藏备用。
1.2.3 基质标准曲线配制 称取不含3种赭曲霉毒素的咖啡空白基质样品,按1.2.2的方法处理得到基质溶液,使用该溶液配制成质量浓度为0.1~20.0 ng/mL的标准工作曲线溶液。
1.2.4 样品前处理 称取1.0 g(精确至0.01 g)咖啡粉碎样品至50 mL干净离心管中,加入100 ng/mL的13C20-OTA内标标准溶液200 μL及10 mL乙腈提取液(V乙腈∶V甲酸∶V水为65∶5∶30),分别漩涡超声5 min,加入3.2 g QuEChERS盐包,剧烈振摇分散,10 000 r/min离心5 min。吸取2 mL上清液上ZanChERS-Myco17净化小柱,收集滤液过0.22 μm PTFE滤膜,上质谱测定。
1.2.5 线性关系 以质量浓度为横坐标、峰面积为纵坐标,将3种赭曲霉毒素系列混合标准曲线溶液分析结果绘制校准曲线,计算相应线性方程及相关系数。
1.2.6 检出限及定量限 通过测定系列混合标准曲线溶液,检出限以3倍信噪比(S/N=3)时对应的目标物浓度换算,定量限以10倍信噪比(S/N=10)时对应的目标物浓度换算。
1.2.7 回收率和精密度测定 分别选取不含3种赭曲霉毒素的咖啡生豆、烘焙咖啡粉、速溶咖啡粉作为基质,分别配制含混标溶液2,5,10 μg/kg 3个水平的供试品溶液进行测定,每个水平6个平行,计算各水平目标物回收率和相对标准偏差。
1.2.8 实际样品测定 采用市售的咖啡生豆(预包装)、咖啡生豆(散装)、研磨咖啡粉(预包装)、研磨咖啡粉(散装)、速溶咖啡(预包装)、速溶咖啡(散装)共30个样品,主要产地为海南省,按试验建立的方法及GB 5009.28—2016进行测定,每种类型5个样品,每个样2个平行。
1.2.9 数据处理 采用SPSS 22.0软件进行数据处理,采用Origin 8.0软件绘图。
2 结果与分析
2.1 质谱条件优化
分别取质量浓度为20 ng/mL的OTA、OTB、OTC和13C20-OTA标准溶液上机调谐,依次选择正、负离子模式扫描一级质谱,比较正、负离子模式的响应情况。结果发现,4种目标物采用正离子模式响应最好。继续采用正离子模式扫描二级质谱,同时选择多反应监测模式优化各目标物的其他参数,筛选最优的定性和定量离子。优化后的质谱参数见表1。
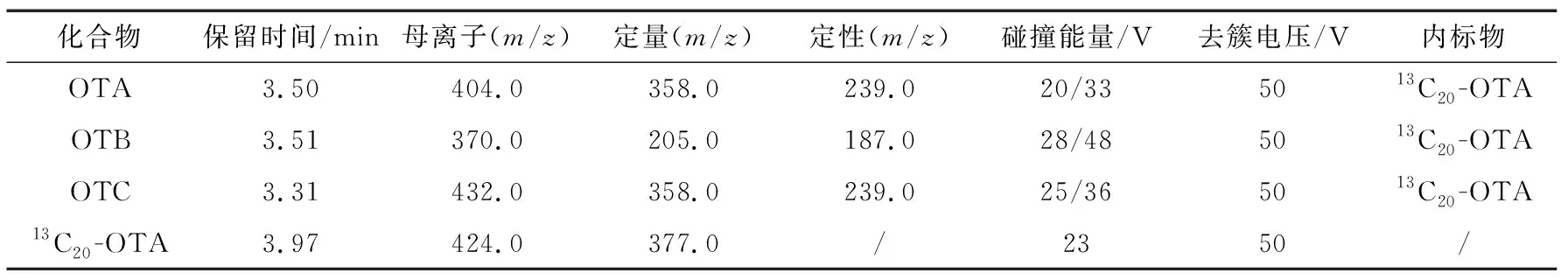
表1 OTA、OTB、OTC和13C20-OTA的质谱参数
2.2 色谱柱及流动相的筛选
试验对比了Waters ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm)、CORTECS T3(2.7 μm,2.1 mm×100 mm)、CORTECS T3(1.8 μm,2.1 mm×100 mm) 3款类型不同填充粒径的色谱柱和0.2%甲酸水/甲醇、水/甲醇、0.2%甲酸水/乙腈、水/乙腈4种不同配比流动相的分离效果。结果显示,采用Waters BEH C18及CORTECS T3色谱柱均能分离4种化合物,其中Waters BEH C18色谱柱响应值最高,分离效果最好。李硕等[24]采用UPLC-MS/MS测定咖啡粉中黄曲霉毒素和杂色曲霉素,发现C18色谱柱填料为实心核壳颗粒分离效果要优于全多孔颗粒。对比4种不同配比流动相,当流动相中添加甲酸后,峰型对称、信号响应更高,并随着甲酸浓度的增加,其响应不断增强。其中0.2%甲酸水/乙腈作为流动相的色谱峰型及信号响应最好。叶林链等[12]研究发现,选择乙腈为流动相,不加酸,OTA、OTB的保留时间会发生漂移,加酸后能改善目标化合物的峰型。赭曲霉毒素化合物由于含较多羧酸,添加适量的酸可以使其保持分子形式有利于色谱柱的保留,促进分离[25]。因此,选择Waters BEH C18色谱柱,0.2%甲酸水/乙腈为流动相。4种化合物质谱总离子流图如图1所示。
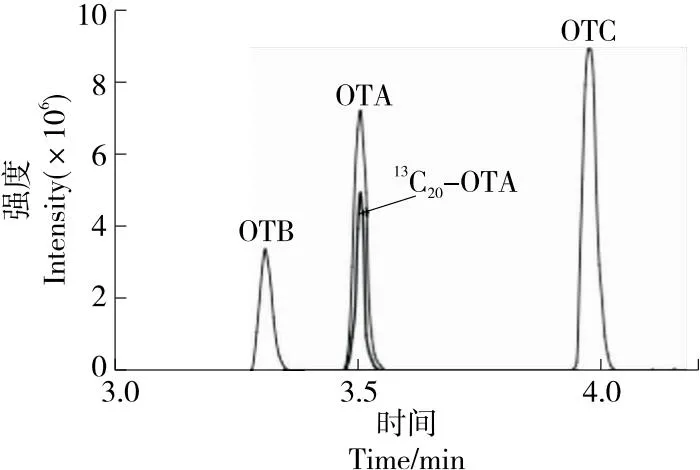
图1 OTA、OTB、OTC和13C20-OTA质谱总离子流图
2.3 样品前处理条件优化
乙腈作为提取溶剂具有沉淀蛋白的作用,对脂肪及蛋白含量较高的食品具有较好的选择[26]。通过加标回收试验考察了6种不同配比提取溶剂1[乙腈—水(V乙腈∶V水为90∶10)]、提取溶剂2[乙腈—水(V乙腈∶V水为75∶25)]、提取溶剂3[乙腈—水(V乙腈∶V水为65∶35)]、提取溶剂4[乙腈—水(V乙腈∶V水为55∶45)]、提取溶剂5[乙腈—水—甲酸(V乙腈∶V水∶V甲酸为55∶40∶5)]、提取溶剂6[乙腈—水—甲酸(V乙腈∶V水∶V甲酸为55∶35∶10)]的提取效果。由图2可知,溶剂中加入适量水可以增强乙腈的渗透性,添加无机盐可以促进乙腈与水相分层,当V乙腈∶V水为65∶35时,开始分层,其中V乙腈∶V水为55∶45的分层效果最好,提取效率最高。当提取液中含有5%甲酸时,OTA、OTB的回收率较高,但甲酸含量过高,3种目标化合物的回收率均降低。有研究[27]表明,体系中加入适当的酸可增强对酸敏感的真菌毒素的提取效果并降低基质效应。试验中加入适当的酸及无机盐可以使目标物保留分子形式利于提取及减少乙腈中水溶性杂质的基质效应,从而获得较高提取效果。综合考虑,选择乙腈—水—甲酸(V乙腈∶V水∶V甲酸为55∶40∶5)作为提取溶剂。
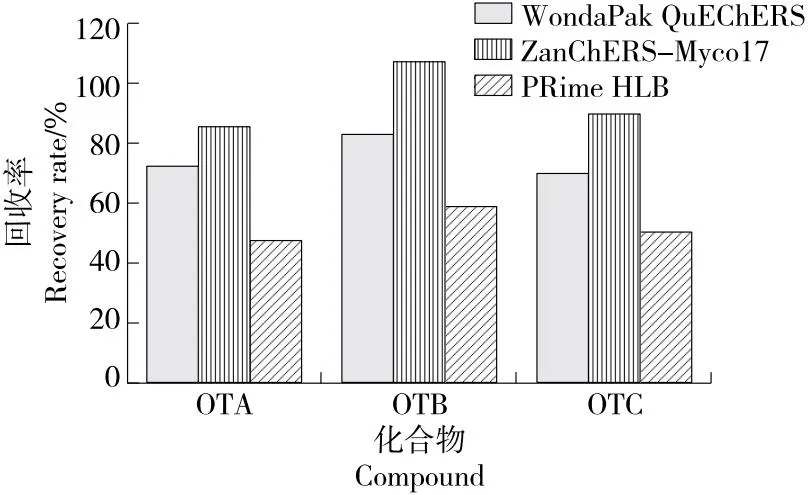
图2 提取溶剂对3种化合物回收率的影响
2.4 净化方式优化
研究[28]表明,可采用不同类型净化方式去除干扰物质,提高净化效果。试验分别选用WondaPak QuEChERS净化管、ZanChERS-Myco17净化小柱、PRime HLB净化柱3种净化方式,以回收率为指标,考察3种目标化合物的净化情况。由图3可知,WondaPak QuEChERS净化管、ZanChERS-Myco17净化小柱对3种目标化合物净化效果较好,其中ZanChERS-Myco17净化小柱净化效果最佳,整体回收率为85.5%~107.5%。QuEChERS净化管采用的净化剂含有PSA、GCB、C18,这些成分可有效去除脂类、糖类及色素等成分,但会对酸性及含有苯环的官能团毒素具有吸附作用,可能是导致回收率偏低的原因。PRime HLB净化柱能够有效去除蛋白、盐、磷脂等干扰物,但对咖啡色素的净化效果不明显,基质干扰大,导致回收率偏低。ZanChERS-Myco17净化小柱采用磁性金属有机骨架(MOFs)材料键合NH2基团,相比净化管具有较高的理论塔板数及选择性。因此,选取ZanChERS-Myco17净化方式。
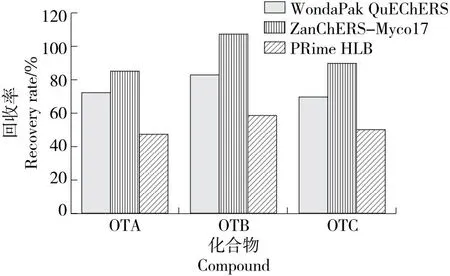
图3 净化方式对3种目标化合物回收率的影响
2.5 线性范围、检出限及定量限
García-Moraleja等[29]以生咖啡豆为基质,建立了UPLC-MS/MS测定OTA的方法,检出限及定量限分别为1.13,1.45 μg/kg。由表2可知,试验方法在0.1~20.0 ng/mL的质量浓度范围内3种目标物线性关系良好,相关系数均>0.999 46,检出限为0.1~0.2 μg/kg,定量限为0.2~0.7 μg/kg,其中OTA的检出限及定量限分别为0.2,0.7 μg/kg,均小于同类方法及GB 5009.96—2016中的检出限和定量限,说明试验方法的灵敏度较高。

表2 3种化合物的线性关系、检出限、定量限
2.6 回收率与精密度试验
基质效应是影响检测结果准确性的重要因素,减少基质效应的影响一般可选择同位素内标法和基质匹配校正法[30-31]。鉴于咖啡基质较为复杂,试验选择内标法定量。分别选取不含目标物的咖啡生豆、研磨咖啡粉、速溶咖啡粉作为阴性样品,各添加2,5,10 μg/kg 3个水平浓度OTA、OTB和OTC混标溶液及内标溶液,按1.2.7的方法分别计算3种化合物的平均回收率及相对标准偏差(RSD),每个浓度6个平行,结果见表3。由表3可知,3种毒素的平均回收率为79.0%~103.0%,RSD为1.5%~7.6%,方法准确性和重现性较好,满足检测要求。

表3 3种化合物的回收率和RSD
2.7 实际样品测定
由表4可知,30份咖啡样品中共检出4批含有OTA,含量为0.82~2.15 μg/kg,均低于标准规定限值(5.0 μg/kg)。经比对,除样品YMS4因检测结果低于标准方法定量限而无法准确定量外,试验方法测定结果与标准方法的相对标准偏差均<15%,说明该方法测定样品结果准确可靠。一般来说,咖啡样品中赭曲霉毒素的污染主要来自生产、运输、贮藏过程中残留毒素的直接接触及真菌侵染后产生代谢物两个方面,此次检出的4批样品中,3批来自散装贮藏,可能是由于散装咖啡密封不严、贮藏条件控制不当而被真菌污染产生毒素所致。生咖啡豆中OTA残留量通常会比烘焙咖啡的高,主要原因是烘焙过程中高温可一定程度上降低OTA水平[32-33]。试验也发现,生咖啡豆OTA检出含量要高于研磨咖啡。
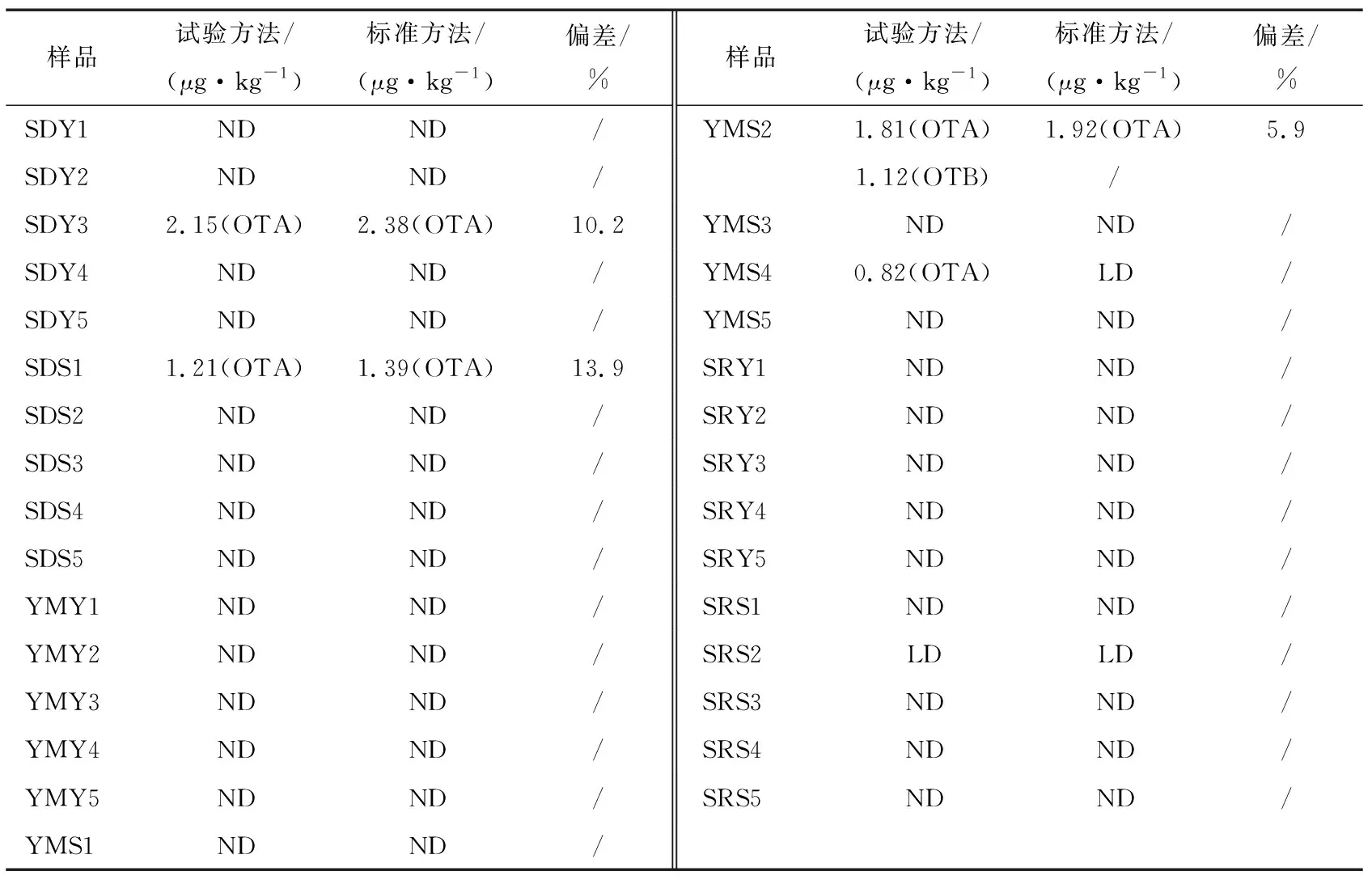
表4 实际样品检测结果†
相关研究[34]数据显示,咖啡及制品在一些国家或地区具有较高的赭曲霉毒素检出率及残留量。对实际样品检测发现,赭曲霉毒素检出率及含量均较低,另外,散装贮藏方式咖啡检出率高于预包装,说明可能存在真菌污染导致毒素积累的风险。建议在咖啡生产及销售过程中应对贮藏条件及方式加以严格控制,防止产毒真菌污染。
3 结论
采用QuEChERS前处理方法结合内标法定量,通过优化色谱质谱条件、提取条件和净化条件,建立了可同时测定咖啡基质中3种赭曲霉毒素的UPLC-MS/MS方法。该方法相较标准方法的免疫亲和柱法更加简便、准确、灵敏,其分析时间短,适用于咖啡基质中3种赭曲霉毒素的快速筛查及定量检测。后续可结合微生物学手段明确污染源,以期为全面监测、评估咖啡中赭曲霉毒素的污染风险提供依据。
