Differential expression of LncRNA and bioinformatics analysis of serum microvesicles in patients with polycystic ovary syndrome
2023-12-28,,,,
, , , ,
(1.Department of Reproductive Medicine, Department of Obstetrics and Gynecology, Affiliated Hospital of Zunyi Medical University, Zunyi Guizhou 563099, China; 2.Key Laboratory of Maternal &Child Health and Exposure Science of Guizhou Higher Education Institutes, Zunyi Guizhou 563099, China)
[Abstract] Objective To determine the different expression profiles of long noncoding RNAs (lncRNAs) in serum microvesicles from patients with polycystic ovary syndrome (PCOS) and controls and to analyze their biological functions. Methods Sixty-five serum samples were collected from each of the PCOS patients and controls, and microvesicles were extracted by differential centrifugation and subjected to high-throughput sequencing to screen out differentially expressed lncRNAs (P<0.05). qRT-PCR was performed to validate nine of these differential lncRNAs. Gene Ontology (GO) functional annotation analysis of cis-regulatory coding genes associated with differentially expressed lncRNAs, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis and reactome pathway enrichment analysis were used to analyze the biological functions of lncRNAs in PCOS serum microvesicles. Results A total of 5 410 lncRNAs were identified in human serum microvesicles of the PCOS and control groups, and a total of 736 differentially expressed lncRNAs were identified, of which 374 were upregulated and 362 were downregulated. The qRT-PCR was performed to validate 9 differential lncRNAs, and the results showed that 5 lncRNAs showed the same trend as the sequencing results. The pathway enrichment analysis showed that these molecules may play an important role in cell proliferation and apoptosis, inflammation, and adipogenesis. Conclusion In this study, serum microvesicles lncRNAs expression profiles of women with PCOS were found to be different from those of control women. Microvesicles may play a role in the pathogenesis of PCOS as carriers of pathogenic lncRNAs.
[Key words] microvesicles;polycystic ovary syndrome;lncRNA;bioinformatics;high-throughput sequencing
Polycystic ovary syndrome (PCOS) is the most common reproductive endocrine disorder affecting the metabolism and reproductive system of women in their reproductive years, with a prevalence of 6.1% to 19.9%[1-2]. Currently, the primary cause of anovulatory infertility is PCOS[3]. PCOS patients usually present with ovulatory dysfunction, clinical and biochemical hyperandrogenism, and polycystic ovarian morphology. This condition is closely related to infertility, insulin resistance, diabetes, obesity, cardiovascular disease, and cancer, and many patients with PCOS suffer from anxiety and depression[4], seriously harming women’s physical and mental health[5-6]. At present, the pathogenesis of PCOS is still unclear, and exploring the pathogenesis of PCOS has become a research hotspot in the field of reproductive medicine.
Microvesicles (MVs) are one type of extracellular vesicles (EVs), and large extracellular vesicles (IEVs) have a diameter of approximately 100~1 000 nm[7]. Formed by cell membrane budding and shedding[8], MVs can selectively send directional instructions to nearby or distant cells and, as an important cell communication mode, regulate their functions and phenotypes[9-11]. MVs contain nucleic acids (DNA and RNA), such as mRNA, long noncoding RNA (lncRNA), small interfering RNA (siRNA), microRNAs (miRNAs), and membrane proteins and lipids from the cell surface[12]. MVs can participate in a variety of pathological and physiological processes, such as apoptosis, autophagy, immune response, tumor growth, and metastasis[13]. Studies have shown that the RNA carried by MVs may play an important role in gene regulation[14]and is closely related to the pathological process of many diseases.
LncRNA is one of the various RNAs found in MVs. LncRNA is an RNA molecule composed of more than 200 nucleotides. This molecule plays an important role in epigenetic regulation, gene transcription, protein translation, and mRNA turnover and is related to the occurrence and development of a variety of diseases. These conditions include inflammation, cancer, autoimmune disease, cardiovascular disease[15], psychiatric disease[16], diabetes[17]and PCOS[18]. Geng et al[19]found that lnc-MAP3K13-7∶1 could inhibit granulosa cell proliferation and promote the development of PCOS. Zhang et al[20]found that MALAT1 sponges miR-125b and miR-203a and regulates TGFβ signaling, thereby contributing to the pathophysiological process of PCOS. However, there are relatively few studies on MVs and MVs derived lncRNAs in PCOS, and there are certain limitations. Based on this, we speculate that female serum MVs in PCOS can serve as carriers of pathogenic lncRNAs, and the abnormal expression of lncRNAs may play an important role in the pathophysiological process of PCOS, and its mechanism deserves further research.
1 Materials and methods
1.1 Research subjects Infertile patients (<35 years old) who received assisted reproductive technology in the Reproductive Center of the Affiliated Hospital of Zunyi Medical University from September 2021 to April 2022 and from September to October 2023 were selected as the research subjects. PCOS infertility patients (n=65) were selected as the PCOS group (experimental group). The inclusion criteria were based on the Rotterdam criteria[21], and non-PCOS patients with clinical manifestations similar to PCOS were excluded. The control group included 65 women with a normal menstrual cycle and regular ovulation and female tubal factor infertility patients (n=65). Patients were excluded from the two groups due to primary hypothalamic amenorrhea, primary ovarian failure, prolactinopathy and other organic and functional diseases of the hypothalamic pituitary ovarian axis. Endocrine diseases such as Cushing’s syndrome, hyperprolactinemia, acromegaly, congenital adrenal hyperplasia, thyroid disease, and androgen-secreting tumors, reproductive system diseases such as endometriosis, history of ovarian surgery or radiotherapy and chemotherapy, genetic diseases such as idiopathic hirsutism, and drug-induced hyperandrogenism, as well as infertility caused by male factors, were also used. This study was approved by the Ethics Committee of Zunyi Medical University(NO:KLLY-2021-195), and all patients signed informed consent before the study.
1.2 Main reagents and instruments A BCA protein quantification kit (Jiangsu Biyuntian Biotechnology Co., Ltd.); PVDF membrane (Millipore, USA); SDS-PAGE loading buffer (Shanghai Ya Enzyme Biomedical Technology Co, Ltd); mouse anti-CD63 monoclonal antibody (Novus, USA); recombinant rabbit monoclonal GAPDH antibody (Hangzhou Hua’an Biotechnology Co, Ltd); SDS (Shanghai Ya Enzyme Biomedical Technology Co, Ltd); a JEM1400Flash transmission electron microscope (Japan Electronics); a ChemiDoc-Touch chemiluminescence imaging system, enzyme standardization instrument (Bio-Rad, Inc, USA); Fluorescence quantitative PCR instrument (CFX series); gradient PCR instrument (T100) and micro spectrophotometer (Nano-500) were used.
1.3 Extraction of MVs The serum samples of women with PCOS and healthy control women were collected from the Department of Reproductive Medicine, Affiliated Hospital of Zunyi Medical University and centrifuged at 2 000×g for 10 min. The supernatant was taken, centrifuged at 3 000×g for 20 min to remove cells and cell debris, aspirated into disposable sterile de-enzymatic centrifuge tubes and stored at -80 ℃ for 2 hours in the refrigerator. Equal amounts of serum were taken from both groups. The supernatant was thawed in a constant temperature water bath at 25 ℃ and placed on ice, diluted 3 times with precooled phosphate-buffered saline (1×PBS, pH 7.4) at 4 ℃ and centrifuged at 500×g for 10 min, 2 000×g for 15 min and 5 000×g for 15 min in a high-speed centrifuge. The supernatant was retained, removed by centrifugation at 20 000×g for 1 h, and washed twice with 1 ml of PBS. The wall of the tube was washed twice, and the samples was transferred to a 1.5 ml sterile nonenzymatic centrifuge tube and centrifuged again at 20 000×g for 1 h. The precipitate was kept, resuspended in 100 μl of PBS, and stored at -80 ℃.
1.4 Identification of MVs For identification of the isolated MVs, transmission electron microscopy (TEM) was used to observe the MV morphology, Western blotting was used to detect the expression of the MVs marker molecule CD63, and a nanoparticle tracking analyzer was used to measure the concentration particle size of MVs.
MVs samples were thawed on ice, lightly blown and mixed at room temperature; then, 20 μl drops were placed on a copper net, excess liquid was aspirated with absorbent paper and left to dry for 2 min, negative dye (30 μl of 3% phosphotungstic acid solution) was stained for 5 min, the sides of the filter paper were blotted dry and this procedure was repeated three times after pure water drops were blotted dry. After drying, the copper net was placed in a sample box and observed under a transmission electron microscope, and images were obtained.
Detection of microvesicular protein expression by protein blotting (Western blot) The total protein of each group of MVs was extracted, and 20 μg protein samples were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred, blocked, incubated with primary antibody dilution CD63 (1∶2 000) overnight at 4 ℃ and secondary antibody dilution (1∶5 000) for 1 hour, washed with TBST and exposed, and the PVDF membranes were developed and analyzed by a Bio-Rad chemiluminescence imaging system.
1.5 Extraction, isolation and sequencing of MVs RNA Total RNA was extracted from the samples by the TRIzol method, and the RNA samples were assessed using Qubit 3.0 and an Agilent 2100 Bioanalyzer. After quality control, libraries were constructed using the KAPA RNA HyperPrep Kit with RiboErase (HMR) for the Illumina library building kit. The designed DNA probes were first hybridized with the RNA samples, thus removing the rRNA from the total RNA, followed by fragmentation of the RNA and synthesis of the first strand of cDNA using the strand-specific method. The second strand cDNA was labeled with dUTP during synthesis, and end repair was completed in this step, followed by A-tail addition, ligation, ligation product purification and fragment size sorting, library amplification, etc. After amplification, the RNA-Seq library was recovered with magnetic beads. After library construction, the libraries were initially quantified by Qubit 3.0, followed by detection of the libraries by an Agilent 2100 Bioanalyzer, and the effective concentration of the inserted target fragments was accurately quantified by qPCR (effective library concentration>3 nM). After the reads were filtered to obtain high-quality data (clean data) and compared with the reference genome (mapping), with TMM (trimmed mean of M-values) normalization and filtering of lncRNAs (counts per million CPM>1), we used the LNCCipedia database to analyze long-stranded noncoding RNA; fold change 1.5,P<0.05, and FDR<1 indicate a significant difference. After obtaining the data, we used FastQC software for quality assessment. Then, the reads were processed for cutting and removal of low-quality sequences, and the quality was qualified and compared to the genome for comparative quality control using Bowtie2 software, followed by the analysis of sample composition using linear algebraic calculations.
1.6 LncRNAs reverse transcription and real-time quantitative PCR
1.6.1 LncRNAs reverse transcription lncRNAs reverse transcription was performed using the PrimeScript TMRT reagent Kit (Perfect Real Time) reagent kit. The reverse transcription system was prepared as follows: 1 μl RNA sample, 2 μl 5*PrimeScript Buffer, 0.5 μl PrimeScript RT Enzyme Mix I, 0.5 μl Oligo dT Primer, 0.5 μl Random 6 mers, 5.5 μl RNase Free dH2O, reaction conditions: 37 ℃ for 15 minutes, 85 ℃ for 5 seconds.
1.6.2 Quantitative PCR of lncRNAs Obtaining lncRNAs sequences through high-throughput sequencing and designing lncRNAs Specific primers. The primer dry powder was centrifuged at 4 000 rpm at room temperature for 5 minutes, and DEPC water was added. After thorough mixing and centrifugation, a primer working solution with a concentration of 10 μm was prepared, and stored for a short time at 4 ℃. The sequence of lncRNAs and internal reference primers is as follows(Table 1).Prepare a quantitative PCR system as follows: 2×SYBR Green PCR mix (12.5 μl), Forward Primer (1 μl), Reverse Primer (1 μl), Template DNA (1 μl), DEPC created Water (DNase, RNase free) (9.5 μl).

Table 1 Primers and sequences for RT-PCR reaction
Reaction conditions: Pre denaturation at 95 ℃ for 10 minutes, followed by 40 cycles of amplification, including 10 seconds at 95 ℃, 60 ℃ for 30 seconds. According to previous literature reports, GAPDH was used as the internal reference for quantitative PCR of microbubble lncRNA[22]. Quantitative PCR results are obtained using formula 2-ΔΔctPerform calculations.
1.7 Bioinformatics analysis of lncRNAs MVs Heatmaps and volcano maps were drawn based on the results of differential expression analysis to obtain the proximity of clustering between samples. The cis-acting target genes (cis-gene) were predicted based on the principle that lncRNAs function is correlated with neighboring coding protein genes, which are usually the coding genes closest to the lncRNAs. We then used the R software clusterProfiler to perform Gene Ontology (GO) functional annotation analysis of cis-regulated coding genes (cis-regulated genes) of differentially expressed transcripts and Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome pathway enrichment analysis to infer the potential biological functions of differentially expressed lncRNAs, withPvalues<0.05 as the criterion for a significant difference.
1.8 Statistical analysis Fisher’s exact test was used to assess the biological function of the statistically significant overlap between differentially expressed gene sets and functional gene sets (e.g., GO/KEGG). LncRNA quantitative PCR data were calculated using Equation 2-ΔΔct, and normally distributed variables were analyzed byttest to determine statistical significance.Pvalues<0.05 were considered to indicate statistically significant differences.
2 Results
2.1 Morphology of MVs isolated from human serum
The TEM results showed that the MVs were round vesicles with a typical bilayer membrane structure (Fig 1A,B). The Western blot results showed that CD63(28 kDa), a microvesicular marker molecule, was detected in the obtained human blood MVs (Fig1C). These results confirmed that the extraction of human blood MVs was successful.

A,B: The electron microscopy images of the extracted MVs show a cup-shaped structure with a diameter of approximately 90-500 nanometers. Scale bar: 200 nanometers;A for PCOS group,B for Control group; C: Western blotting showed the presence of CD63 protein in MVs. Fig 1 Identification diagram of extracted human serum MVs
2.2 Principal component analysis (PCA) PCA uses a linear algebraic computational method to reduce the dimensionality and extract the principal components of hundreds of genetic variables, where x and y represent the two main principal components, and the samples between the two groups were well separated, indicating that the obtained results are of high quality and are acceptable for subsequent result analysis, as shown in Fig 2.

A: Principal component analysis of samples, Exp for PCOS group, ctrl for control group; B: Principal component analysis of the first 500 most changed features, dim is the dimension. Fig 2 Principal component analysis of MVs samples
2.3 Differential lncRNA expression analysis and cluster analysis The gene expression (read count) was obtained by analyzing the read comparison results with RSEM software, followed by TMM normalization and filtering of lncRNAs, and the results are shown in (Fig 3A-B), with count per million(CPM)>1, indicating that genes with sufficient abundance were obtained for subsequent operations.
In this study, a total of 106959 lncRNA transcripts were identified in all samples, and 5 410 lncRNA transcripts were normalized and filtered by TMM. A total of 736 differentially expressed lncRNA transcripts were significantly expressed: 374 upregulated and 362 downregulated transcripts. In addition, volcano plots and heatmaps were plotted based on the results of differential expression analysis (Fig 3C-D) to visually demonstrate the distribution of differential gene expression between the two groups of samples and the clustering of distant and close relationships between samples. The top 10 lncRNAs with the most significant up and downregulation are listed in Table 2,Fig 3 shows the volcano plot and heatmap of differentially expressed lncRNAs.
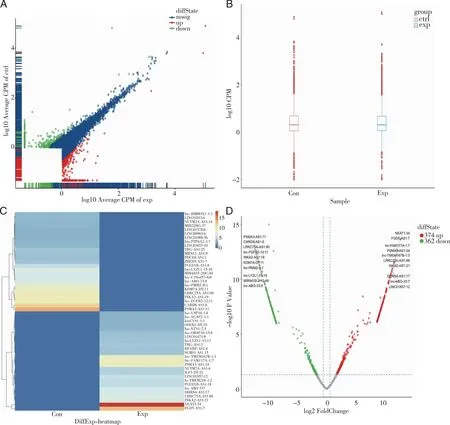
A: Analysis of differential gene expression in the PCOS and control groups; B: normalized gene expression, exp is the experimental group, ctrl is the control group;C: heatmap of differentially expressed lncRNAs, horizontal coordinates indicate samples, vertical coordinates indicate differentially expressed lncRNAs, red indicates high expression, and green indicates low expression;D: volcano plot of differentially expressed lncRNAs,Top10 diffExp-volcanoplot.Fig 3 Differentially expressed lncRNAs in MVs of PCOS patients and non-PCOS patients

Table 2 Differentially expressed lncRNAs
Fold change is presented as lncRNA expression in women with versus without polycystic ovary syndrome (PCOS); up indicates upregulation, down indicates downregulation.
2.4 qRT-PCR validation of lncRNA results in MVs Subsequently, we utilized qRT-PCR to validate the differentially expressed 9 lncRNAs in the sequencing results in 20 women with PCOS and 20 women with control. Nine lncRNAs were differentially expressed in the sequencing results. 6 of these lncRNAs were differentially expressed between PCOS patients and controls and were consistent with the trend of the sequencing results, of which the up-regulated ones in PCOS women were NEAT1∶34(P=0.244), FGD5-AS1∶7(P=0.171) and PSMA3-AS1∶24(P=0.757), and the down-regulated ones were INKA2-AS1∶19(P=0.042) and CARD8-AS1∶6(P=0.883). of which INKA2-AS1∶19(P<0.05) was statistically significant. there was an opposite trend for LNC-TMEM167B-1∶3(P=0.276) and PSMA3-AS1∶11(P=0.097), and no significant difference for LNC-FAM117A-1∶7(P=0.906) and LRRC75A-AS1∶90(P=0.466,Fig 4).

Exp is the PCOS group, ctrl is the control group;*:P<0.05.Fig 4 Serum LncRNA qRT-PCR validation results microvesicles
2.5 Functional analysis of differentially expressed lncRNAs For the differentially expressed lncRNAs transcripts, GO pathway enrichment analysis, KEGG functional annotation analysis and Reactome pathway enrichment analysis were performed on their cis-regulated genes to identify the potential biological functions of the differentially expressed lncRNAs. We conducted GO analysis from three aspects(Fig 5): biological process, cellular component and molecular function. The results showed that the main categories were regulation of behavior, positive regulation of telomerase activity, mitotic cell cycle arrest, epidermal growth factor receptor binding. PCOS is correlated with positive regulation of telomerase activity, mitotic cell cycle arrest, and epidermal growth factor receptor binding.

Annotated graph of GO functions of cis-regulated coding genes in differentially expressed lncRNAs, Y-axis: GO-enriched entries, X-axis: gene ratio. Fig 5 GO enrichment analysis of related coding genes in lncRNAs differentially expressed in MVs of PCOS patients and non-PCOS patients
KEGG pathway results mainly showed enrichment in biosynthesis of cofactors and glycerophospholipid metabolism, glycosphingolipid biosynthesis-lacto and neolacto series, nicotinate and nicotinamide metabolism, glycosphingolipid biosynthesis and ganglio series, etc (Fig 6A-B). Reactome pathway results showed that they were mainly enriched in transcriptional regulation by TP53 and glycerophospholipid biosynthesis, transcriptional regulation of white adipocyte differentiation, and MAPK6/MAPK4 signaling (Fig 6C-D).

A: Plot of KEGG pathway enrichment analysis of cis-regulated coding genes in differential lncRNAs, X-axis: Gene number, Y-axis: KEGG enrichment entries; B:X-axis: Gene Ratio, Y-axis indicates KEGG enrichment entries; C: Plot of Reactome pathway enrichment analysis, X-axis: Gene number, Y-axis: Reactome enrichment entries; D: X-axis: Gene Ratio, Y-axis: Reactome enrichment entries, -log10(p) denotes significance. Fig 6 KEGG and Reactome enrichment analysis of related coding genes in lncRNAs differentially expressed in MVs of PCOS patients and non-PCOS patients
3 Discussion
PCOS is a common complex endocrine disease in women of reproductive age. Its symptoms and signs include hyperandrogenism (clinical manifestations include hirsutism and/or acne), polycystic ovary morphology, and ovulation dysfunction (decreased ovulation/anovulation)[23], which not only affects fertility function but also causes endocrine and metabolic dysfunction[24], the specific mechanism of which has not yet been fully elucidated. MVs, as a novel information mediator, can participate in the physiological and pathological processes of organisms, and MVs are involved in the regulation of biological functions by wrapping and delivering genetic material and signaling molecules (including miRNA, lncRNA, circRNA and proteins, etc), which are involved in regulating the biological functions of neighboring and distant receptor cells[25]. As one of the new etiological research directions of PCOS, lncRNAs have received much attention in recent years. As the important roles of MVs and lncRNAs in various diseases are being explored, we focused on the role of MVs and the lncRNAs they carry in the development of PCOS.
In this study, human serum MVs lncRNAs were mainly used as the research object, and the morphology and surface-specific protein molecules of MVs were identified by TEM and Western blotting. After detection, the isolated extracts were found to be consistent with the description of MVs morphology and characteristics in the literature[26]. We obtained the expression profiles of serum microvesicular lncRNAs by high-throughput sequencing and obtained 736 significantly differentially expressed lncRNAs. A total of 374 of them were upregulated, and 362 were downregulated. After performing qRT-PCR validation, we discovered that out of all the lncRNAs that matched the sequencing results, NEAT1, FGD5-AS1∶7 and PSMA3-AS1∶24 exhibited a subtle increase, while INKA2-AS1∶19 and CARD8-AS1∶6 demonstrated a decline. INKA2-AS1∶19 was statistically significant, but the validation outcomes were not statistically conclusive. Among these 9 lncRNAs, NEAT1 has been reported to be associated with PCOS, previous studies have found that NEAT1 mediates the miR-324-3p/BRD3 axis to alleviate metabolic disorders and ovarian pathological changes in mice with PCOS[27]. Our results of MVs-derived NEAT1 are consistent with this finding and may be involved in the pathological process of PCOS to a certain extent. The other lncRNAs verified are mainly reported to be related to tumor growth, migration, and invasion[28-29]. Currently, there is a lack of research related to PCOS or its complications. The exact role of these lncRNAs in the pathogenesis and progression of PCOS still needs to be elucidated. Some of the differentially expressed lncRNAs we identified have also been reported to be associated with PCOS. MVs-derived TUG 1 was highly expressed in this study. Previous studies have found that overexpression of TUG 1 leads to excessive follicle activation and growth and interferes with dominant follicle selection by regulating granulosa cell proliferation and steroidogenesis[30]. MVs-derived TUG 1 may regulate granulosa cell proliferation to a certain extent, leading to follicular development and ovulation disorder in PCOS. Downregulation of MALAT1 in granulosa cells may be involved in the pathophysiological process of PCOS by regulating the TGFβ signaling pathway[20]. Some studies have also shown that downregulation of MALAT1 aggravates PCOS by regulating miR-302d-3p-mediated leukemia inhibitory factor activity[31], and our results are the same. These results suggest that the lncRNAs in serum MVs of the PCOS group in this study may be involved in the pathogenesis of PCOS.
To further explore the potential mechanisms of MVs-derived differentially expressed lncRNAs for PCOS, we performed GO, KEGG and Reactome pathway enrichment analysis to clarify their cis-regulated coding gene. We performed GO, KEGG and Reactome pathway enrichment analysis to identify the pathways that cis-regulate the enrichment of coding genes and thus determine the potential biological functions of differentially expressed lncRNAs. A class of biological molecules that play an important role in cell metabolism and physiological function are cofactors, nicotinamide adenine dinucleotide (NAD+) is an important cofactor in various biological processes[32], and the reduction of mitochondrial dysfunction in granulosa cells of PCOS patients is related to the high level of NAD+[33]. The results of our pathway enrichment analysis showed that lncRNAs were enriched in the cofactor biosynthetic pathway, suggesting that our lncRNAs may mediate PCOS development by acting on signaling molecules in the cofactor biosynthetic pathway.
The pathway enrichment results also showed that the differentially expressed lncRNAs were associated with glycerophospholipid metabolism. Interestingly, it has been shown that abnormal expression of LPCAT1 is associated with dysregulated lipid metabolism in PCOS[34]. Moreover, dysfunctional glycerophospholipid metabolism in the follicles of PCOS patients was shown to play an important role in the IVF process[35]. The results suggested that lncRNAs in PCOS serum MVs may also be associated with signaling molecules in the glycerophospholipid metabolic pathway mediating the onset of impaired lipid metabolism in women with PCOS.
Glycosphingo liposomes (GSLs) are components of mammalian cell membranes and are involved in regulating various aspects of cell biology, including cell proliferation, apoptosis and inflammation[36]. GSL is subdivided into structural series such as gangliose-, lactose-/neolactose-, globose-, and heterogeneous-series, and abnormal expression of glycosphingolipids has been reported in many diseases such as cancer, neurocardiovascular diseases, lipid storage diseases, metabolic syndrome, and PCOS[35, 37-38]. The cis-regulated coding genes of differentially expressed lncRNAs in this study were enriched in the glycosphingolipid biosynthesis ganglio series, glycosphingolipid biosynthesis - globo and isoglobo series pathways, suggesting that PCOS serum microvesicular lncRNAs may mediate the regulation of cell biological processes through sphingolipid-related pathways, leading to the development of PCOS-related abnormalities in lipid metabolism, inflammation, and cellular value-added or apoptosis.
The mitogen-activated protein kinase (MAPK) signaling pathway is a major regulatory module for various cellular processes, such as cell proliferation, differentiation and stress response[39]. Previous studies have found that mitogen-activated protein kinase 4 and phosphorylated ERK1/2 are reduced in PCOS granulosa cells and that these pathways may affect granulosa cell function in PCOS[40]. The differentially expressed genes in this study were enriched in this complex, suggesting that PCOS serum microvesicular lncRNAs may mediate PCOS granulosa cell function and differentiation through the MAPK signaling pathway.
The results of the present study suggest that differentially expressed lncRNAs do exist in human serum MVs from PCOS patients, but studies on the expression of lncRNAs in human serum MVs and their pathways of action are still limited. Given the limitations of the small sample size in this study, future studies with expanded sample sizes are necessary to elucidate the molecular mechanisms of these differentially expressed lncRNAs.
In this study, we found that the serum microvesicular lncRNAs expression profiles of women with PCOS differed from those of healthy women. MVs may act as carriers of pathogenic lncRNA and play a role in the pathogenesis of PCOS.
