RUNX1在Ang II 诱导心肌肥厚中的作用及机制
2023-12-28丁立群王玉玖杨丽娟沈嘉祺邹明锐刘宝辉
丁立群 王玉玖 杨丽娟 沈嘉祺 王 傲 邹明锐 刘宝辉*
1 滨州医学院 山东 烟台 264003;2 滨州医学院附属医院 山东 滨州 256603
心肌肥厚是一种在血流动力学压力负荷增加下的代偿反应,通过增加心肌总量、加强心肌收缩力等使机体维持正常的血液循环,同时有一定的储备力。但是这种代偿能力也有不利之处,肥厚心肌的需氧与供血不能平衡,造成心肌缺血、心肌收缩力减退。这种供需不匹配进一步导致了多种心血管疾病的发生,包括心肌梗死、心力衰竭甚至心源性猝死[1]。因此,心肌肥厚被认为是心血管疾病发病和死亡的预测因子,其发病机制和防治的研究是心血管领域的研究热点。
Runt相关转录因子1(runt-related transcription factor 1,RUNX1)是转录因子核心结合因子家族的成员之一,在造血、成骨和神经发生中起着重要作用,对其他发育过程也很重要[2]。对RUNX1的研究重点集中在血液疾病和癌症上,而近年来越来越多的研究指出RUNX1在心血管疾病中也发挥着重要作用[3]。但RUNX1在心肌肥厚中的作用却罕有报告。本研究旨在探讨RUNX1对血管紧张素Ⅱ(angiotensin Ⅱ,Ang Ⅱ)诱导的心肌肥厚的作用及机制。
1 材料与方法
1.1 细胞培养与实验处理 H9C2细胞购自武汉普诺赛生命科技有限公司。收到细胞后在37℃、5%CO2的细胞培养箱中培养,适应24 h后胰酶消化传代。细胞培养基为10%胎牛血清、1%青霉素链霉素溶液的DMEM高糖培养液。根据处理条件的不同,分为以下8组,分别为正常对照组(Control组),10 nM Ang II处理组(L-Ang II组),100 nM Ang II处理组(M-Ang II组)[4],1 000 nM Ang II处理组(H-Ang II组),慢病毒转染无义序列(NS)组,慢病毒转染无义序列+Ang II处理(Ang II+NS)组,慢病毒转染沉默RUNX1(shRUNX1)组,慢病毒转染沉默RUNX1+Ang II处理(Ang II+shRUNX1)组。以上组别的细胞均在无血清培养基饥饿24 h后换为完全培养基,加用不同浓度的Ang II或PBS处理48 h。包装好的慢病毒购自于山东维真生物科技有限公司,根据说明书加入慢病毒原液并使用ADV-HR助感染,使RUNX1基因在H9C2细胞中沉默,嘌呤霉素筛选稳转株,转染72 h后收集细胞进行下一步处理分析。
1.2 Western blot检测 新配裂解液(RIPA裂解液:蛋白酶抑制剂=100∶1),加入收集的H9C2细胞沉淀中,吹打混匀后冰上裂解40 min,再4℃、12 000×g离心10 min,小心吸取上清液,BCA法测量蛋白浓度,加入4×Loading buffer,在100℃下热变性5 min,根据蛋白浓度调整每孔上样量为30 μg,通过10% SDS-PAGE电泳分离蛋白质样品,再将蛋白转至PVDF膜上。室温下脱脂牛奶封闭1 h,然后在4℃下孵育一抗过夜。所用一抗为:RUNX1抗体(BOSTER,51 kDa),稀释比为1∶500;RUNX1抗体(Proteintech,51 kDa),稀释比为1∶500;SERCA2a抗体(BOSTER,115 kDa),稀释比为1:1 000;GAPDH抗体(BOSTER,37 kDa),稀释比为1∶7 000。再加二抗室温下孵育1 h。用ECL显影液及伯乐Bio-Rad凝胶成像系统(Gel DocTM XR+)对膜上的条带进行可视化并用Image J软件进行定量评估。实验重复三次取均值。
1.3 PCR分析 用普洛麦格总RNA提取试剂盒(LS1040)提取H9C2细胞沉淀的总RNA,并用赛默飞K1622试剂盒进行反转录。为了量化不同mRNA的水平,通过使用以下引物对产物进行qPCR分析:GAPDH引物对为5′-GGCACAGTCAAGGCTGAGAATG-3′和5′-ATGGTGGTGAAGACGCCAGTA-3′,BNP引物对为5′-CAGAAGCTTGGAGCTGATAAG-3′和5′-TGTAGGGCCTTGGTCCTTTG-3′。qPCR条件为:使用NovoStart SYBR qPCR SuperMix Plus试剂盒按厂商说明书配制20 μL体系,程序为95℃ 1 min,95℃ 20 s,55℃ 30 s,72℃ 30 s,循环数为35。
1.4 细胞免疫荧光 室温下将已爬好的细胞玻片放入4%多聚甲醛固定并用0.5%TritonX-100通透细胞膜,滴加正常山羊血清室温下封闭30 min,加入稀释好的一抗(β-actin 1∶100,BOSTER)4℃过夜,孵育完成后在PBST中浸洗,加入FITC标记荧光二抗(BOSTER)37℃湿盒避光孵育1 h。后续加入DAPI染液染核并用含抗荧光淬灭剂的封片液封片,置于倒置荧光显微镜下观察,Image J软件对图像荧光度进行量化处理。

2 结果
2.1 Ang II处理H9C2心肌细胞建立肥厚模型 为了构建体外心肌肥厚模型,本研究在对H9C2细胞饥饿24 h后,加用Ang II(100 nM)处理48 h,并通过检测细胞形态大小及肥厚标记物的表达来判断是否成功建立心肌肥厚模型。如图1所示,对胞内 β-actin蛋白行免疫荧光染色,观察细胞形态及横截面积大小,发现Ang II组的细胞横截面积较Control组增大(P<0.05),细胞形态由原来的长梭形变短、变圆。BNP是心肌肥厚标记物之一,qRT-PCR实验结果提示Ang II组的BNP mRNA表达量较Control组明显升高(P<0.001)。以上结果验证了心股肥厚模型的成功建立。

A.免疫荧光法观察Ang II处理H9C2细胞后细胞形态及横截面积大小的变化,Control组为完全培养基培养,Ang II组为加用100 nM的Ang II处理;B.Image J软件量化分析上述两组细胞的荧光面积,比较两组细胞横截面积大小;C.qRT-PCR检测上述两组心肌肥厚标记物BNP mRNA的表达水平。*P<0.05,***P<0.001。
2.2 RUNX1蛋白在心肌肥厚中的表达 为研究RUNX1基因在心肌肥厚中的作用,本研究收集细胞沉淀进行Western blot检测RUNX1蛋白表达水平。如图2所示,与Control组相比,用Ang II处理组RUNX1蛋白表达水平均出现上调,且随着Ang II用药浓度的增加,RUNX1蛋白的表达量也增加,呈现剂量—效应关系。根据这一结果,猜想RUNX1的表达对心肌肥厚的发生发展具有促进作用,为验证这一猜想,本研究使用慢病毒转染沉默RUNX1基因进行后续实验。

**P<0.01,****P<0.0001。
2.3 沉默RUNX1减轻Ang II诱导的心肌肥厚 为证实RUNX1对心肌肥厚的影响,沉默RUNX1后进一步评价心肌肥厚的两个指标如下:一是细胞的形态与横截面积大小,二是心肌肥厚标记物BNP的mRNA水平变化。慢病毒转染细胞后Western blot检测RUNX1的表达,可见shRUNX1组的RUNX1蛋白表达水平显著下降(图3D),证实慢病毒转染细胞沉默RUNX1成功。如图3C所示,NS组与shRUNX1组的BNP mRNA表达未见明显统计学差异,在加用Ang II处理48 h后,无论是否沉默RUNX1的表达,BNP mRNA的表达水平均较前升高,但是沉默RUNX1的细胞BNP mRNA表达水平升高幅度较小,且Ang II+shRUNX1组较Ang II+NS组的表达量低(P<0.01)。另外,免疫荧光法观察计算细胞横截面积发现,正常表达RUNX1的细胞在Ang II处理后细胞横截面积增加(P<0.05),但沉默RUNX1的细胞加用Ang II处理后细胞横截面积未有明显变化,与正常对照组无明显统计学差异(图3A、3B)。以上两个指标的结果基本一致,证明了沉默RUNX1可以减轻Ang II诱导的心肌肥厚。
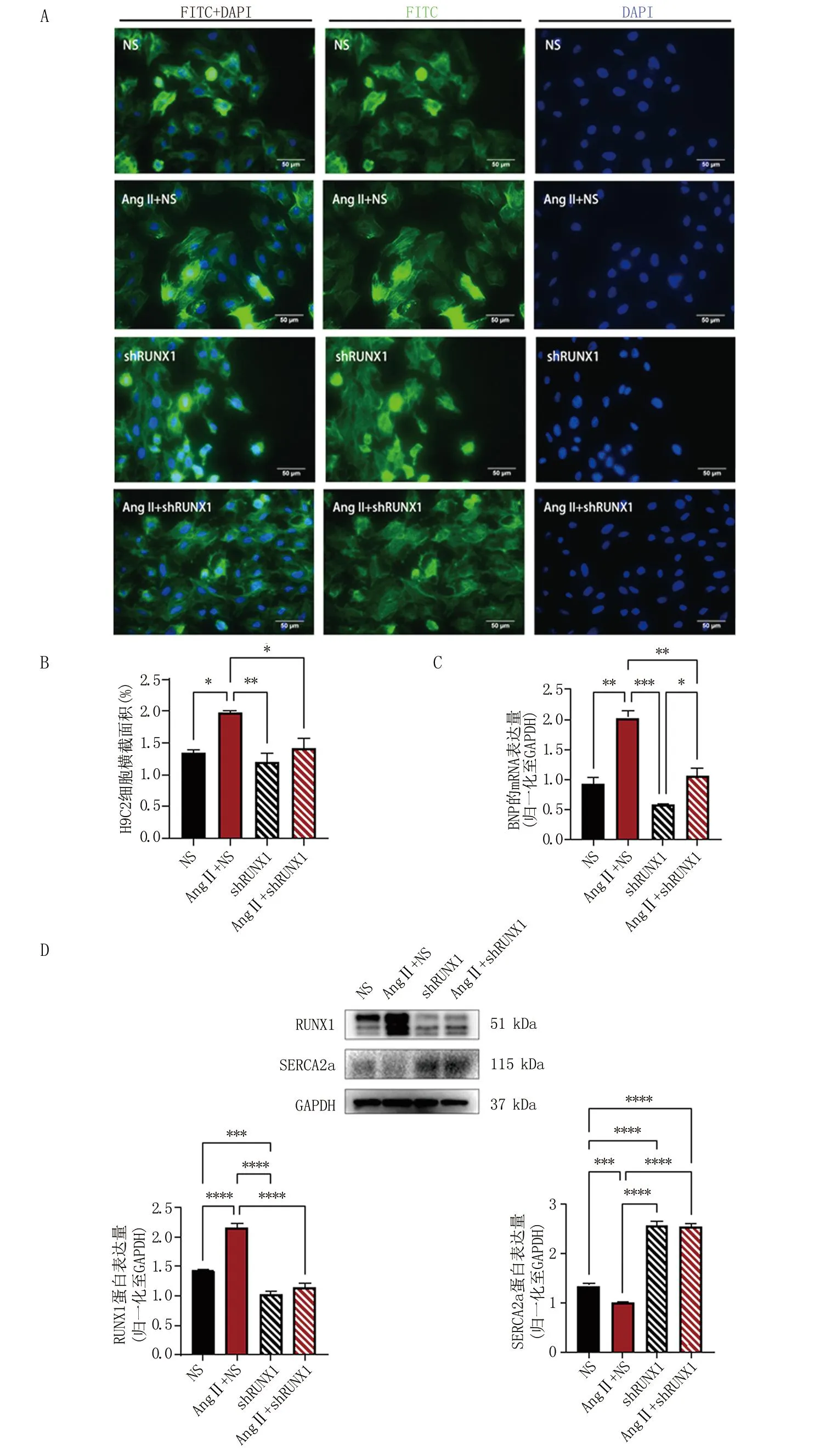
A.免疫荧光法观察Ang II处理H9C2细胞后细胞形态及横截面积大小的变化;B.Image J软件量化分析上述四组细胞的荧光面积,比较四组细胞的横截面积大小;C.qRT-PCR检测上述四组细胞心肌肥厚标记物BNP mRNA的表达水平;D.Western blot分析上述4组细胞SERCA2a、RUNX1蛋白的表达水平。*P<0.05,**P<0.01,***P<0.001。
另外,SERCA2a作为维持细胞钙稳态的重要因子之一,在Ang II+NS组中的表达量较NS组明显降低(图3D)。同时,本研究观察到无论加用Ang II处理与否,沉默RUNX1后心肌细胞的SERCA2a蛋白表达水平均上调。
3 讨论
病理性心肌肥厚是心力衰竭的前期病变,是脑卒中、冠心病、心衰、猝死的独立危险因素,是多种心血管疾病的共同病理变化。此外,病理性心肌肥厚能够导致心功能受损,是心力衰竭和心源性猝死的主要预测因子。因此,阐明心肌肥厚发生的机制,探讨延缓其发生发展的方法对于临床改善心功能有重要意义。研究表明,Ang Ⅱ、内皮素和肾上腺素受体激动剂等多种因素均会导致病理性心肌肥厚的发生[5-6]。本研究用Ang Ⅱ建立心肌肥厚的细胞模型,并发现随着Ang Ⅱ用药浓度的增加,RUNX1的表达量较前明显升高,并呈剂量依赖关系。
RUNX1作为一种祖细胞标记物,在成人心肌组织中表达很低[7],在出生7 d内,心脏的增殖和再生能力丧失的同时,RUNX1基因因甲基化而失活[8]。RUNX1不仅在人体正常发育过程中起到重要作用,也是心脏疾病发生的重要靶点。RUNX1在心肌梗死区及交界区内表达均有升高[9],这一结果不仅出现于啮齿动物的心肌梗死模型中,在心肌梗死患者中也呈现同样的结果[10],降低RUNX1功能可保留心脏收缩力并预防不利的心室重塑[3],然而RUNX1在心肌肥厚发展中的作用并未得到明确阐述。为探讨RUNX1是否作用于心肌肥厚的发生发展,本研究应用Ang II干预慢病毒转染沉默RUNX1的H9C2细胞。从BNP的mRNA表达量检测来看,与shRUNX1组相比,Ang II+shRUNX1组的BNP mRNA表达量虽升高(P<0.05),但升高幅度小于NS组与Ang II+NS组(P<0.01);在静息状态下即未加用Ang II处理时,NS组与shRUNX1组的BNP mRNA表达量未见明显统计学差异,说明单纯的沉默RUNX1基因表达对心肌细胞的面积无影响。从免疫荧光的结果来看,Ang II+shRUNX1组较Ang II+NS组的细胞横截面积小(P<0.05),此结果与BNP mRNA的变化情况一致,说明沉默心肌细胞内RUNX1表达可明显改善Ang II诱导的心肌肥厚,进一步提示RUNX1参与心肌肥厚的病理过程。
在体内外心肌细胞上,各种刺激如牵拉、Ang II、ET-1及儿茶酚胺等均可使胞内的Ca2+浓度升高。应用Ryanodine预先耗尽胞内Ca2+贮库可抑制牵拉及Ang II等的心肌肥厚反应。SERCA2a作为维持细胞内钙稳态重要的因子,通过调节肌质网(sarcoplasmic reticulum,SR)的钙摄取来调控心肌细胞的收缩与舒张。McCarroll等人[3]研究发现沉默RUNX1对SR功能的影响是小鼠心梗后所观察到的有益作用的主要贡献者。本研究发现,Ang II可明显降低心肌细胞内SERCA2a的表达;而沉默心肌细胞内RUNX1后可明显上调由Ang II导致的SERCA2a表达量的降低。结果表明,RUNX1可能通过抑制SERCA2a参与心肌肥厚过程。
本研究还存在一定的局限性。仅在体外进行实验验证 RUNX1可以下调SERCA2a的表达参与心肌肥厚的病理过程,并未从体内模型出发探讨RUNX1的整体作用效果。另外对SERCA2a的研究也仅仅是在蛋白水平,缺乏转录水平及蛋白活性的验证,未能验证SERCA2a的表达在RUNX1介导心肌肥厚中的作用。以上有待今后进一步研究明确。
综上所述,RUNX1在心肌细胞中表达,并在Ang II诱导的心肌肥厚模型中显著上调。RUNX1对心肌肥厚能够通过抑制SERCA2a的表达来参与心肌肥厚的病理过程。本研究结果为心肌肥厚及心衰防治的新干预靶点深入研究提供依据。
