掺杂对Al(111)面氧分子吸附性能影响
2023-12-15张亚婷任克亮纪华杨佳丁丽宏马海萍
张亚婷, 任克亮, 纪华, 杨佳, 丁丽宏, 马海萍
(宁夏大学物理与电子电气工程学院, 银川 750021)
铝是自然界中最常见的金属材料,由于其具有密度小、导电导热性能优异、强度高、耐蚀性强等优点,所以在电子信息、仪器仪表和新能源电池等领域被广泛应用[1-2]。在自然环境中,铝原子和环境中的氧分子在铝金属表面相互作用容易形成一层致密的氧化物薄膜覆盖在金属表面[3-8],从而抑制金属表面发生化学或电化学反应的作用,使其表现出很强的耐蚀性[9-10]。
近年来,随着现代工业技术的发展及重大工程对材料性能的特殊要求,人们通过实验验证,在铝基体中添加其他元素可以有效提升金属铝的性能。刘萍[11]研究了合金元素对7000系铝合金性能及微观组织的影响。何亚玲等[12]研究了A7N01-T4性能随合金元素含量的变化规律。车海亮等[13]研究发现Sm、Er稀土元素掺杂会提高铝合金的热力学性能。陈铁等[14]研究发现合金元素Mn、Mg、Si对3104铝合金组织和力学性能会产生影响。以上研究成果对提升铝合金的力学性能具有重要意义。但由于添加元素对环境介质的敏感性[15],使得材料表面容易发生化学或电化学反应,影响氧原子与铝原子的相互作用,改变了金属表面氧化膜的组织结构,降低了材料的耐蚀性[16]。为了探究添加元素对金属铝表面氧化膜形成机理的影响,人们开展了大量关于掺杂对铝吸附水、氧、以及氢分子影响研究,Wang等[17]研究了氢气在纯铝与Ti掺杂Al(111)表面解离扩散,得到Ti处于次表层时更利于氢原子迁移。陈莉莉[18]、Lu等[19]研究了Si、C、P+掺杂对铝团簇吸附氧分子的影响。Qiao等[20]使用第一性原理研究了O2在Cu、Ag、W掺杂的Al(111)面的吸附。赵剑英[21]研究了Fe、Zn、Cu等掺杂铝团簇吸附水分子的过程,发现Fe掺杂时水的分解能垒最低。郑萌萌[22]使用第一性原理方法研究得到较高的半径和电负性是Sc、Ti、Zr等过渡金属加速铝氢化的主要原因。范立华等[23]研究了Sc、V、Fe、Ti等元素掺杂Al(111)面催化分解H2的过程,发现V掺杂结构催化氢分子分解的效果要优于Sc、Ti等其他掺杂表面。
目前,关于掺杂影响铝合金表面氧化膜形成机理的研究中,涉及Mn、Ni、Si掺杂对铝吸附O2影响的研究较少,为了揭示Mn、Si、Ni合金元素在不同氧原子覆盖度下对金属铝表面形成氧化保护膜的影响规律。现采用基于密度泛函理论(density functional theory, DFT)的第一性原理计算方法,通过模型建立和计算参数的设置,计算不同氧原子覆盖度下Mn、Si、Ni掺杂对Al(111)面吸附O2的吸附能、功函、Bader电荷、差分电荷密度以及态密度的影响。分析掺杂对Al(111)面吸附O2的影响规律,为铝合金材料的防腐性能改善提供理论依据。
1 研究方法和建模
采用基于DFT[24-26]的Materials Studio软件进行计算。计算采用的表面模型为具有12 Å(1 Å=10-10m)真空层的6层(3×3)结构的Al(111)Slab模型,每层有16个原子,图1所示为模型表层。计算中,被吸附物与表面上方4层Al原子可自由弛豫,表面下两层铝原子被固定,使之成为二维无限周期体系。优化过程使用Monkhorst-Pack方法选取网格、将K点设为4×4×1,利用标准算法的电子和离子位置收敛判据为1×10-5eV和3×10-2eV,截断能设置为520 eV。为了研究掺杂对Al(111)面O2吸附性能的影响,在图1所标注阿拉伯数字1、2、3的位置处用Mn、Ni、Si原子替代Al原子,从而形成了含掺杂的结构体系,该体系的吸附能计算公式为
(1)

1、2、3的位置处分别用Mn、Ni、Si原子替代Al原子图1 纯铝和掺杂表面模型Fig.1 Pure aluminum and doped surface model
式(1)中:Ead为吸附能;i为Ni、Si、Mn 3种掺杂元素;N为吸附O2的个数;EAl+i+O2为吸附后表面模型总能量;EAl+i为吸附前模型的总能量;EO2为O2的能量。O2在金属表面吸附的稳定性通过吸附能Ead的绝对值大小表示,绝对值越大,表示氧分子越容易吸附在金属表面,吸附结构越稳定。
针对式(1)中掺杂体系的吸附能与表面模型有关外,还与掺杂元素和O原子覆盖度有关,选取固定O2数量改变掺杂种类和固定掺杂元素改变O原子覆盖度两种方案,分别构建了吸附模型。方案1:在图1各个掺杂表面的掺杂位原子上方垂直距离2 Å处吸附单个O2,并将其初始键长设置为1.23 Å。方案2:利用方案1优化后的模型结果,采用同样的方法,增加表面原子覆盖度,对体系的吸附和电子性质进行计算。
2 掺杂种类对氧分子吸附影响
2.1 吸附计算
为了研究不同掺杂对O2吸附的影响,将Ni、Mn、Si 3种掺杂原子分别放到图1所示的位置,同时平行放置一个O2在掺杂位原子垂直距离2 Å处,通过计算得到了O2解离前后纯铝和掺杂表面的俯视图和侧视图(图2),以及O2解离后O原子间的距离、O原子与掺杂位原子距离、吸附能(表1)。

表1 Al(111)面和掺杂表面吸附能(Ead)、O原子与掺杂位原子距离(dO—M)、O原子距离(dO—O)Table 1 Adsorption energy(Ead) of Al(111) surface and doped surface, distance between oxygen atom and doped atom (dO—M), distance between oxygen atom (dO—O)
通过对图2所示O2吸附前后各表面俯视图及侧视图分析发现,解离前设置O2键长为1.23 Å,解离后各表面O2断键解离成O原子并向各个吸附表面靠近,O原子之间距离增加,各掺杂表面掺杂位原子较吸附前发生了明显的“内陷”。
对表1中O2吸附后O原子间的距离分析得到:与O2在气相状态下键长(1.24 Å)[27-28]相比,吸附后O原子的距离增大至2.67~3.18 Å,说明O2吸附后完全解离转化成为O原子。其中,Mn、Si掺杂时氧原子解离后距离小于纯铝表面,因此,Mn、Si抑制了氧分子解离,而Ni促进了氧分子解离。对比O原子与掺杂位原子解离后的距离发现,O原子与掺杂位Al、Si原子距离小于O2-(1.4 Å)及Al3+(0.53 Å)、Si4+(0.4 Å)的半径之和,O原子和Mn、Ni原子距离略微大于O2-(1.4 Å)与Mn2+(0.67 Å)、Ni2+(0.69 Å)的半径之和;而各表面O原子与最近邻Al原子之间的距离均在1.770~1.905 Å,小于对应离子距离之和。因此,O原子不仅与掺杂位Al、Si原子存在相互作用,还与周围近邻Al原子存在相互作用。
对比Al(111)和掺杂表面吸附能发现,Al(111)和掺杂表面的吸附能均为负值,但纯铝表面的吸附能绝对值最大,说明O2更容易吸附在纯铝表面,解离后的O原子与纯铝表面结合更紧密,形成的吸附结构更稳定。而Si、Ni、Mn掺杂表面吸附能绝对值依次减小,因此掺杂抑制了O2的吸附,吸附解离后O原子与掺杂表面原子相互作用较弱,形成的掺杂表面的吸附结构稳定性降低。
2.2 电子计算
为了研究掺杂种类对铝合金表面氧化膜形成机理的影响和各吸附表面原子之间相互作用,计算了各表面的差分电荷密度(图3)、Bader电荷以及态密度(图4~图6)。

黄色代表电子汇聚,蓝色代表电子散失,掺杂使表面电荷重新排布图3 纯铝及掺杂表面差分电荷密度Fig.3 Differential charge density of pure aluminum and doped surface

图4 O2吸附后各表面O原子及掺杂位原子分态密度图Fig.4 The partial density of states of O atoms and doped atoms on each surface after O2 adsorption
分析图3所示的差分电荷密度,解离后的O原子周围主要呈现为黄色,表现为集聚电子,得到电子;而各表面主要呈现为蓝色,表现为电子散失,失去电子。因此,体系的电子流动方向主要是从各个表面流向O原子,并且主要集中在体系表面。
通过分析Bader电荷中各原子电子得失情况,发现:在各个体系中,掺杂位Al、Si原子以及最近邻Al原子分别失去电子,从而带正电荷;而O原子在各个表面得到电子,从而带负电荷,结果与差分电荷密度结果相符。结合前面的吸附结果得到,氧分子发生解离的原因来自材料表面向氧分子转移了电荷,使氧分子状态发生改变,解离成为了氧离子。结合吸附后原子之间的距离得到氧分子解离后与掺杂位原子之间存在相互作用,其中,硅原子和铝原子之间电荷转移多、距离小,因此,相互作用更强。但是不同掺杂原子失去电子数量与周围铝原子失去电子数量不同,从而原子之间的相互作用也不同,通过计算得到,纯铝表面单个O原子得电子数为1.713个,当Si、Ni、Mn掺杂时,O原子得电子数依次减少:分别为1.681、1.563、1.552个,因此,Mn、Ni、Si掺杂抑制了表面电荷转移,原子间这种电荷转移的原因在于不同原子之间电负性的差异,O原子的电负性大于各个表面原子,当O2吸附时,O原子电负性大,表面原子的电负性小,O原子吸引表面原子的电子,因此得到电子,而各个表面失去电子。因此,单个氧分子吸附条件下,Mn、Ni、Si抑制氧化膜的生成主要是由于掺杂位原子与氧原子之间存在电荷转移,阻碍了Al(111)面向氧分子转移电子,氧化膜形成速度减缓,对金属的保护能力降低。因此,金属的抗腐蚀能力减弱。
图4、图5分别为各表面氧原子、掺杂位原子分态密度以及最近邻铝原子在-10~4 eV区间中电子轨道态密度图,通过分析发现,吸附后纯铝和Si掺杂表面掺杂位原子与O原子之间的态密度趋势极为相似,各原子态密度峰值重叠区域多、共振频率高,最近邻Al原子在费米能级附近态密度峰值个数分布更密集,因此,在纯铝和Si掺杂表面,O原子与掺杂位原子以及最近邻Al原子之间存在很强的相互作用,形成的吸附表面稳定性强、吸附更紧密。在图5(b)中,Mn掺杂导致最近邻Al原子态密度峰值在费米能级附近(-2~2 eV)个数减少使O原子与最近邻Al原子态密度峰值重叠数减少,相互作用减弱,氧化膜形成受阻,而O原子与Mn原子在费米能级附近态密度趋势相差较大,存在共振但共振数目较纯铝和硅掺杂表面明显减少,因此,O原子与Mn掺杂表面之间相互作用弱于O原子与其他表面掺杂位原子;Ni掺杂后,最近邻Al原子费米能级处的态密度峰值个数少于纯铝表面,但较Mn掺杂表面较多,O原子在费米能级附近与Ni原子3d轨道态密度趋势相似。因此,O原子与Ni原子以及最近邻Al原子相互作用较强,虽然可以稳定吸附,但吸附能小于纯铝和Si掺杂表面,O2吸附稳定性较弱。
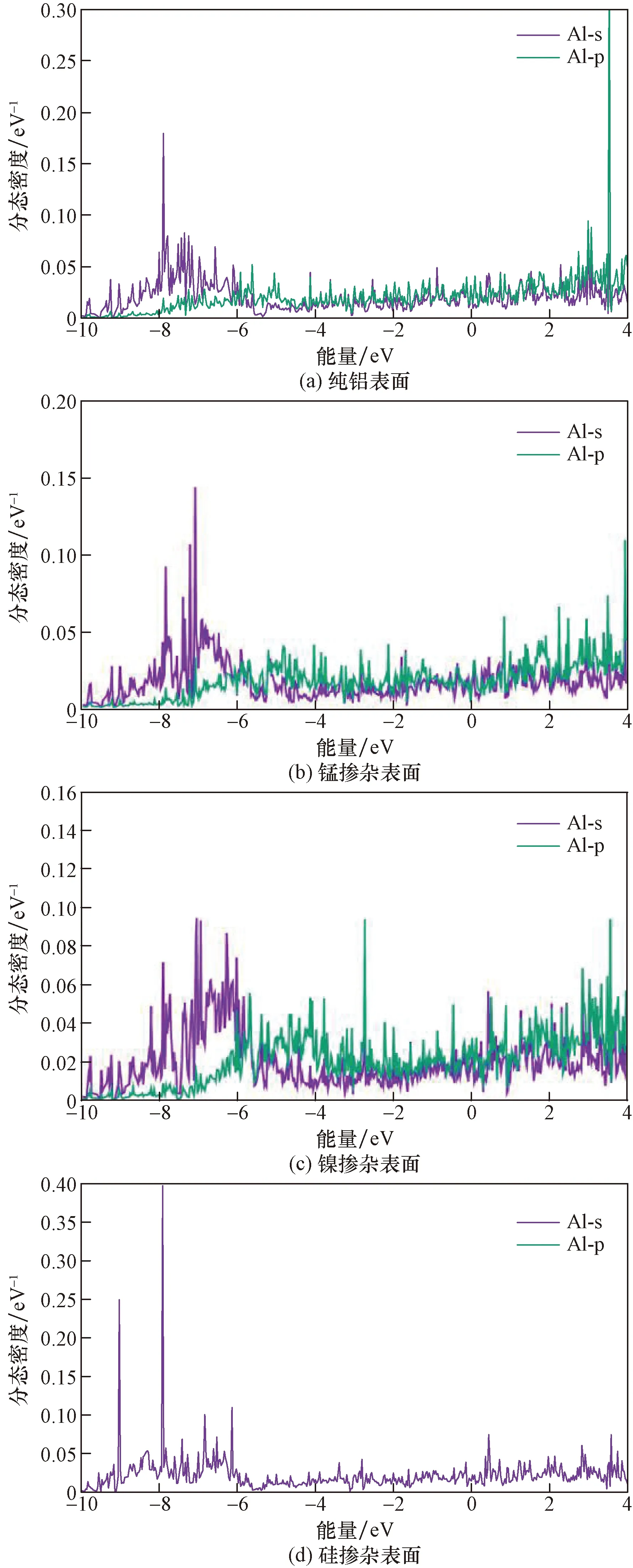
图5 O2吸附后最近邻铝原子分态密度图Fig.5 Partial density of states of nearest neighbor aluminum atoms after O2 adsorption
图6给出了4种吸附表面总态密度-10~4 eV区间内分布情况。分析发现,在整个区间内,各个表面态密度峰密集,证明了氧分子在纯铝表面吸附时电子非常活跃,但各个表面态密度峰值分布区间仍然存在微小差异;结合图4分析发现对于纯铝和Si掺杂表面,整个区间内态密度峰主要来自Al、Si和O原子的p轨道,其余内层轨道几乎对态密度没贡献,但在-10~-9 eV区间内,Si原子的s轨道态密度峰值突然增大,并且参与杂化;而Mn、Ni掺杂表面的态密度峰主要来源于Mn、Ni原子的d轨道以及O原子的p轨道,Mn、Ni原子的s、p轨道以及O原子的s轨道几乎没有贡献,不参与杂化。因此,无论是纯铝或者掺杂表面,O2吸附后与表面原子的相互作用主要是由表面原子的最外层电子轨道的性质决定。并且,对于Mn、Ni掺杂表面,在整个区间内,它们的d轨道主要在高能区参与杂化,O原子的p轨道主要在低能区参与杂化,轨道之间重合范围小,掺杂原子与O原子的杂化弱,相互作用小,而对于纯铝和Si掺杂表面,Al原子的p轨道与O原子的p轨道在-10~4 eV区间内均参与杂化,原子之间相互作用强。同时,结合体系的态密度与之前的Bader电荷得到:氧分子吸附时氧原子与掺杂位原子之间存在电荷转移,因此具有相互作用,这会影响氧化膜的形成,进而影响了金属的耐蚀性。

图6 O2吸附后各表面总态密度Fig.6 The total density of states of each surface after O2 adsorption
3 不同氧原子覆盖度条件下掺杂对Al(111)面吸附性能影响
3.1 吸附计算
除了掺杂元素影响氧化膜形成外,氧原子覆盖度也会影响其吸附性能,覆盖度(Ö)通常是指被吸附的原子个数与表面原子比值[29]。为了研究不同氧分子覆盖度条件下掺杂对Al(111)面吸附性能的影响,在顶位平行吸附方法中,通过添加吸附O2的个数来改变覆盖度,对氧气在各表面的解离情况进行研究。结果发现,当覆盖度达到3/8时,表面弛豫现象明显,有一个铝原子被抬起到表面上方,此时钝化膜的厚度增加;当覆盖度达到4/8时,O原子穿过表层到次表层上方,并与次表层部分铝原子形成化学键,这一结果与杨笑鹤等[30]实验研究结果一致,即当氧化次数增大,O原子会向表面下方延伸。但在各掺杂表面,O原子最终在各表面上方整齐排列,各表面弛豫程度较小,同时得到不同覆盖度O原子条件下纯铝及其掺杂表面吸附O2的吸附能、功函,结果如表2、表3所示。
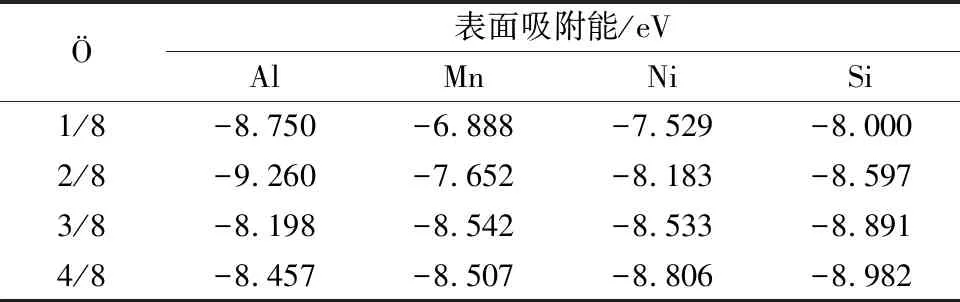
表2 不同O原子覆盖度下各表面吸附能Table 2 Adsorption energy of O atoms with different coverage
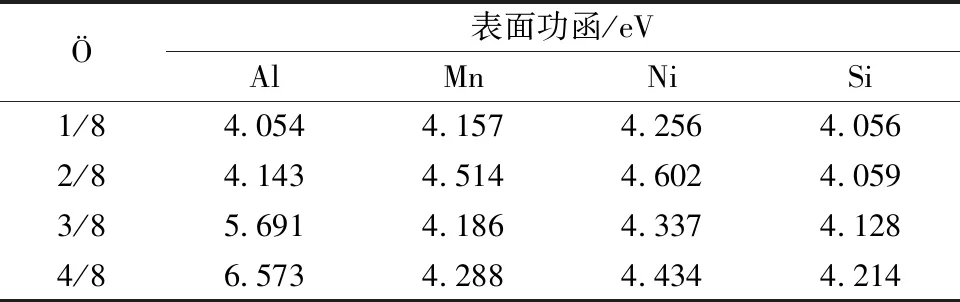
表3 不同O原子覆盖度下各表面功函Table 3 The surface work functions under different coverage of O atoms
通过对比表2中的吸附能发现,Mn、Ni、Si对Al(111)表面吸附O2的影响与O原子的覆盖度有关,当O原子的覆盖度小于2/8时,Mn、Si、Ni掺杂表面吸附能小于纯铝表面。此时,O2在纯铝表面吸附稳定性高于掺杂表面,Mn、Si、Ni掺杂抑制了O2吸附,并且Mn原子掺杂时的吸附能绝对值最小,抑制吸附效果最弱;当覆盖度大于等于3/8时,Mn、Ni、Si掺杂表面吸附能小于纯铝表面,O2在掺杂表面稳定性低于纯铝表面,Mn、Ni、Si掺杂促进了O2吸附,并且Si原子掺杂吸附能最大,促进吸附效果最好。各表面分别考虑时:对于纯铝和Mn掺杂表面,随着覆盖度增大,吸附能绝对值上下波动,因此吸附体系不稳定;在纯铝表面,覆盖度为2/8时吸附能绝对值达到最大值9.26 eV,即吸附两个O2时纯铝吸附结构最稳定,在Mn掺杂条件下,当氧原子覆盖度达到3/8时,吸附能绝对值最大,吸附结构最稳定。对于Si、Ni掺杂表面,随着O原子覆盖度增大,Si、Ni掺杂表面吸附能绝对值增大,形成的吸附结构越稳定。以上结果与钟家淞等[31]、刘雁玲等[32]通过实验研究硅-铝合金随着氧化时间增大,表面没有发生较大差距并且合金的耐蚀性增强的结果一致。对比分析表3中不同条件下的功函值,结果发现:Mn、Ni掺杂表面随着覆盖度的增大,功函上下波动,当O原子覆盖率达到1/4时功函达到最大值,分别为4.514 eV和4.602 eV。而对于纯铝表面和Si掺杂表面,随着表面覆盖度增大,表面功函也呈现增长趋势。
3.2 电子计算
为了进一步研究不同覆盖度条件下掺杂对Al(111)面吸附性能的影响,对各表面不同氧原子覆盖度条件下的差分电荷密度(图7)、Bader电荷(表4),以及吸附后各表面氧原子态密度进行计算(图8~图11)。

表4 不同覆盖度O原子下的单个O原子的Bader电荷Table 4 The Bader charge of a single O atom under different coverage O atoms

图7 不同覆盖度O原子各表面差分电荷密度Fig.7 Differential charge density of O atom with different surface coverage
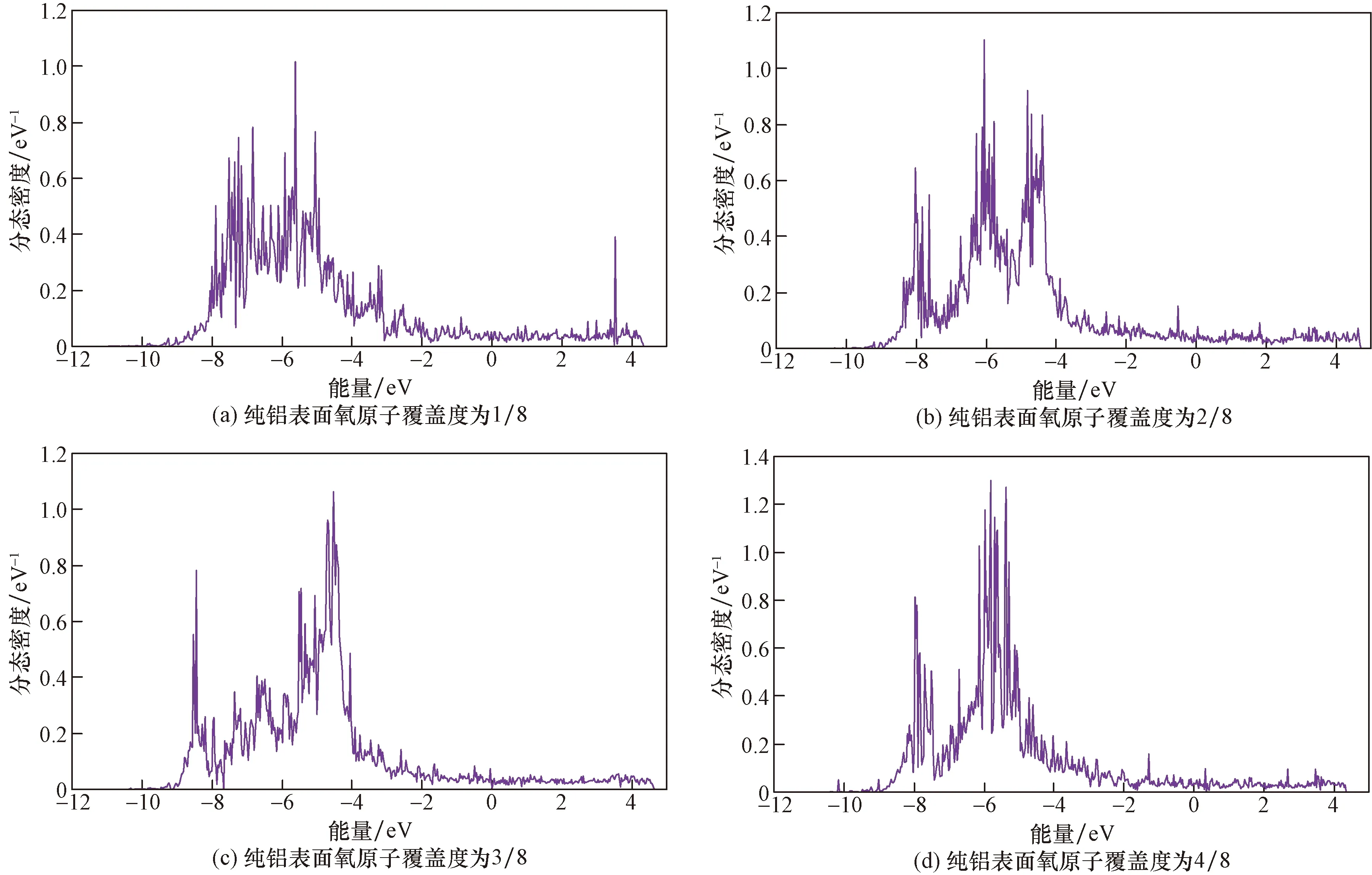
图8 纯铝表面氧原子最外层电子轨道Fig.8 The outermost electron orbital of oxygen atom on pure aluminum surface
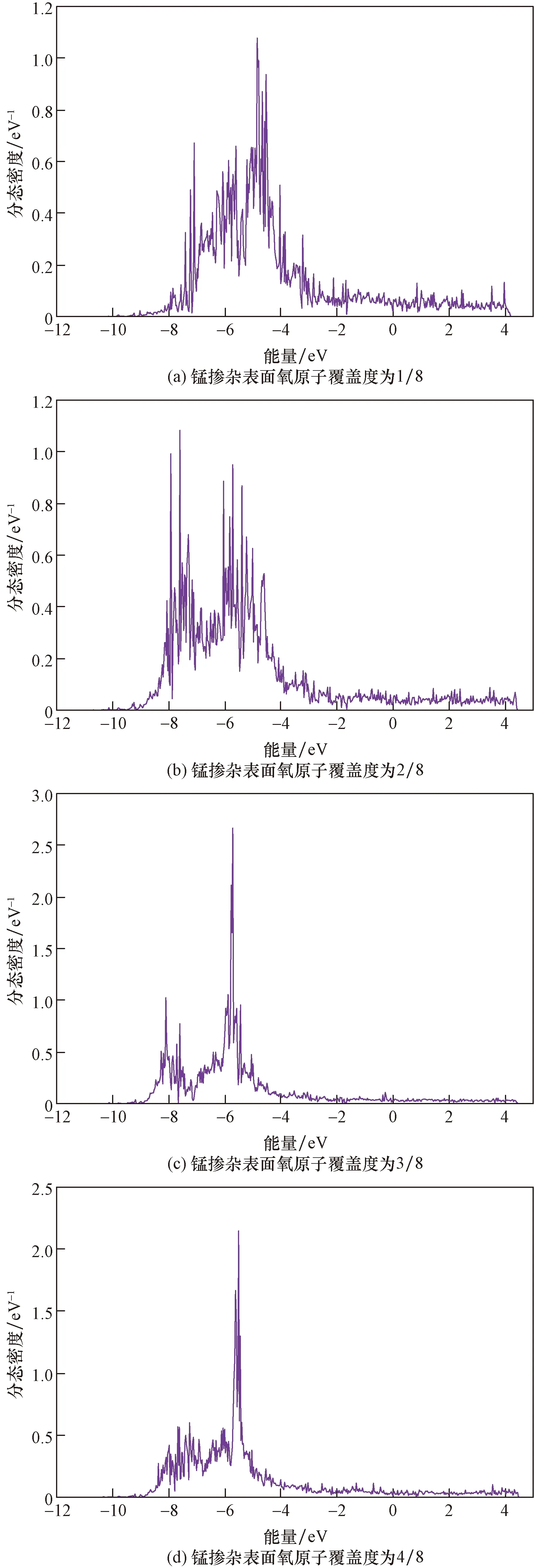
图9 锰掺杂表面氧原子最外层电子轨道Fig.9 The outermost electron orbital of oxygen atom on manganese doped surface

图10 镍掺杂表面氧原子最外层电子轨道Fig.10 The outermost electron orbital of oxygen atom on nickel doped surface

图11 硅掺杂表面氧原子最外层电子轨道Fig.11 The outermost electron orbital of oxygen atom on silicon doped surface
对图7进行分析可知,随着覆盖度增大,各个表面的电子发生重新排布,吸附O原子的差分电荷密度始终呈现黄色,因此,O原子周围始终表现为电子汇聚的地方;而表面各原子呈现蓝色,表现为电子散失,各表面电子主要从表面流向O原子。
分析表4中Bader电荷数,对各个表面不同O原子覆盖度下得电子数对比发现:在Si掺杂表面,随着覆盖度增大,O原子得电子数增大;对纯铝和Mn掺杂表面,当O原子覆盖度增大时,O原子得电子数先增大后减小;而Ni掺杂表面,覆盖度增大,O原子得电子数先减小后增大。另外,对于纯铝以及Mn掺杂表面,O原子得电子数分别在覆盖度为2/8、3/8时最大,此时表面向O原子转移电荷数最多。在同一表面条件下:当O原子覆盖度小于等于2/8时,掺杂表面O原子得电子数小于纯铝表面,此时,Mn、Ni、Si原子抑制了各表面向O原子转移电子,当覆盖度大于2/8时,掺杂表面O原子得电子数大于纯铝表面,此时,Mn、Ni、Si原子促进了Al(111)面向O原子转移电荷,结果与前面的吸附计算规律相一致。最终研究发现,影响体系吸附性能的原因来源于体系与氧分子之间的电荷转移。
图8~图11分别为不同O原子覆盖度下纯铝、锰、镍以及硅掺杂表面氧分子解离后在-12~4 eV区间内的最外层轨道态密度图。比较发现:当O原子覆盖度增大时,对于Mn掺杂表面,随着覆盖度增大,态密度向左移动,能量朝着低能方向进行,体系的稳定性增加,覆盖度达到4/8时,态密度向右移动,体系稳定性下降;而对于Si、Ni掺杂表面,态密度都向左移动,体系的能量朝着低能方向移动,因此,Si、Ni掺杂表面吸附体系的稳定性增加。对于纯铝表面,当覆盖度小于2/8时,态密度先向左移动,此时体系能量朝着低能方向移动,体系稳定性增加,当体系的覆盖度达到3/8时,态密度开始向右移动,能量向着高能方向移动,体系的稳定性降低,结果与计算得到体系吸附能的结果一致。
4 结论
针对掺杂元素和O原子覆盖度对Al(111)吸附O2的影响,采用第一性原理方法,对固定O原子覆盖度改变掺杂种类和固定掺杂元素改变O原子覆盖度两种条件下,Mn、Ni、Si掺杂Al(111)面吸附O2的吸附能、功函、Bader电荷进行计算,对比分析了掺杂表面原子的差分电荷密度和态密度,从不同角度揭示了Mn、Ni、Si掺杂Al(111)面吸附氧气的机理,分析了不同O原子覆盖度时含掺杂表面性质随覆盖度变化规律。
(1)单个O2吸附时,各表面O2均发生解离,因此,Mn、Ni、Si掺杂不影响氧分子的解离行为。
(2)Mn、Ni、Si掺杂对Al(111)吸附影响与O原子的覆盖度有关,当O原子覆盖度小于3/8时,Mn、Si、Ni掺杂抑制O2吸附,这一特性与原子的电子性能有关,是由于掺杂抑制了Al(111)面向氧原子转移电子,影响了氧化膜的形成,从而使金属的抗蚀性能降低。当O原子覆盖度大于等于3/8时,Mn、Si、Ni掺杂促进了O2吸附;此时O原子与近邻Al原子之间的相互作用增强,促进氧化铝形成,增强了金属抗蚀性能。
(3)固定掺杂元素后,Si、Ni掺杂表面随着O原子覆盖度增加,吸附能增大,表面向氧原子转移电子数增多,O2越容易吸附,表面越容易被氧化,促进氧化铝膜形成,纯铝和Mn掺杂表面随氧原子覆盖度增大,吸附能震荡变化,吸附表面不稳定,在特定O原子覆盖度下会促进氧化铝膜形成,从而有效改善金属耐蚀性。
